Crouzon, Apert, Pfeiffer, Saethre-Chotzen, and Carpenter Syndromes
Crouzon Syndrome
Crouzon syndrome was first described in 1912.
Inheritance
Inheritance is autosomal dominant with virtually complete penetrance. It is caused by multiple mutations of the fibroblast growth factor receptor 2 gene, FGFR2. [1, 2, 3]
Features
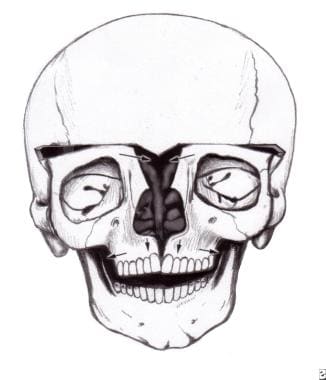
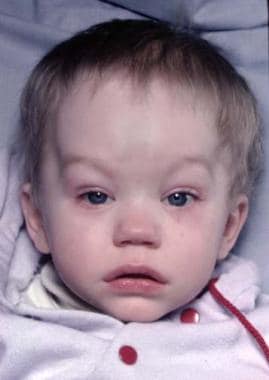
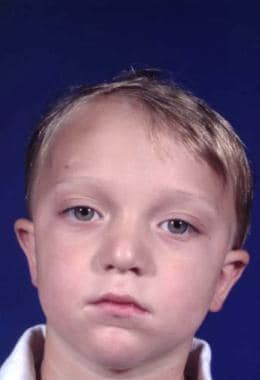
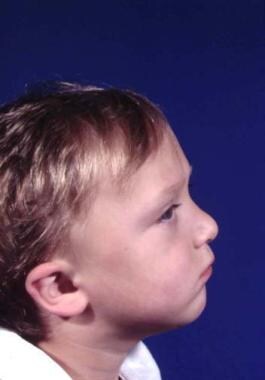
Features of the skull are variable. The skull may have associated brachycephaly, trigonocephaly, or oxycephaly. These occur with premature fusion of sagittal, metopic, or coronal sutures, with the coronal sutures being the most common. In addition, combinations of these deformities may be seen. [4] See the image below.
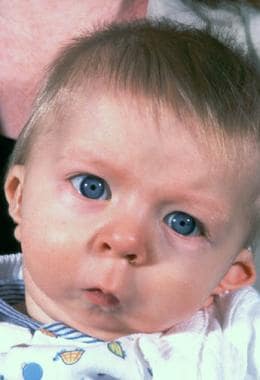
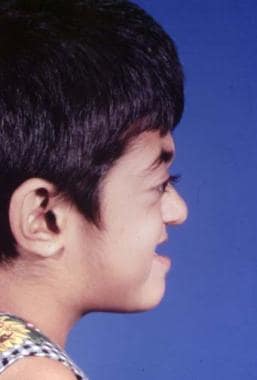
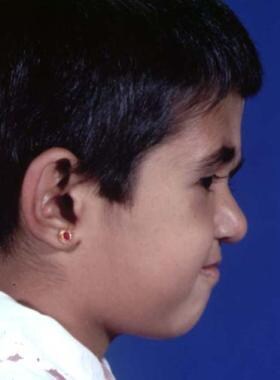
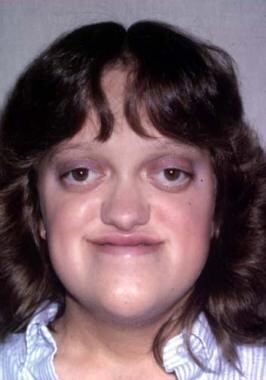 Typical appearance of a patient with Crouzon syndrome, with maxillary retrusion, exorbitism, and pseudoprognathism. Anteroposterior view.
Typical appearance of a patient with Crouzon syndrome, with maxillary retrusion, exorbitism, and pseudoprognathism. Anteroposterior view.
The orbits are shallow with resulting exorbitism, which is due to anterior positioning of the greater wing of the sphenoid. The middle cranial fossa is displaced anteriorly and inferiorly, which further shortens the orbit anteroposteriorly. The maxilla is foreshortened, causing reduction of the orbit anteroposteriorly. All these changes result in considerable reduction of orbital volume and resultant significant exorbitism. In severe cases, the lids may not close completely. The maxilla is hypoplastic in all dimensions and is retruded. This decreases the anteroposterior length of the orbital floor.
A study by Forte et al found that in both Crouzon and Apert syndrome, the bony orbit is shortened, orbital and orbital soft-tissue volumes are reduced, and the globe’s volume is increased. In the study, which included 10 children with Apert syndrome, nine children with Crouzon syndrome, and 12 controls, the length of the bony orbit was 12% and 17% shorter in the Apert and Crouzon syndrome patients, respectively; the bony orbital volume was 21% and 23% smaller, respectively; the globe’s volume was 15% and 36% larger, respectively; and the orbital soft-tissue volume was 19% and 29% less, respectively. [5]
The upper dental arch in Crouzon syndrome is narrowed and retruded, which results in a class III malocclusion. Premature contact of the molars also may be present; this produces an anterior open bite. This may cause the mandible to be rotated downward and backward. The chin and malars are hypoplastic. [6, 7]
Investigations
Examination of the eyes by an ophthalmologist is essential to assess for papilledema, which indicates elevated intracranial pressure. Another finding may be optic atrophy with deterioration of vision; fortunately, this is rare.
Radiologic examination
This consists of standard radiology to produce anteroposterior, lateral, and cephalometric views. The information gained is the position of the maxilla relative to the mandible. This is class III, with the upper teeth lying behind the lower teeth when they are in occlusion. Patients show evidence of elevated intracranial pressure and have "paw marking" of the skull due to the gyri of the brain indenting and thinning the calvaria, with, in severe cases, erosion. The fusion of the involved sutures can be seen. See the image below.
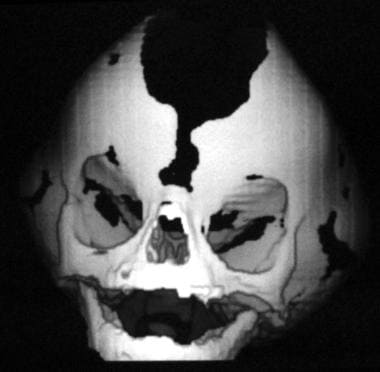 Crouzon syndrome. Anteriorly, the widened frontal and sagittal sutures can be seen.
Crouzon syndrome. Anteriorly, the widened frontal and sagittal sutures can be seen.
A CT scan helps to confirm the findings of standard radiographs and provides information on ventricular size. Three-dimensional CT scans can be produced but yield no more information than standard scans, although suture fusion can be graphically displayed.
General assessment
Other abnormalities are sought, and the child's mental development is carefully assessed. An orthodontist should see the child and initiate treatment when indicated; this is usually preparation for maxillary surgery.
Apert Syndrome
In nearly all patients with Apert syndrome, the cause is 1 of 2 FGFR2 mutations involving amino acids (Ser252Trp, Pro253Arg). The condition is inherited in an autosomal dominant mode. [8]
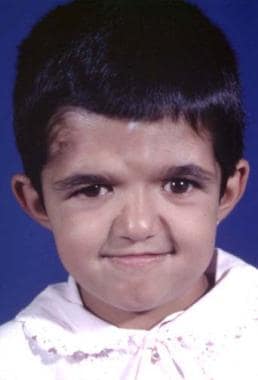
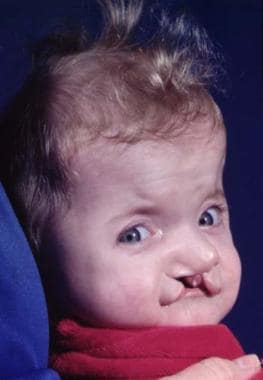
Craniosynostosis is present, characterized by brachycephaly and, frequently, turricephaly; the anterior fontanelle is enlarged. [9] The maxilla is hypoplastic with a high-arched palate, class III malocclusion with an anterior open bite, and, frequently, a cleft of the soft palate. The midface is hypoplastic. This, together with the retrusion, causes exorbitism. Complex syndactyly of the hands and feet is present. It is symmetric, and other limb anomalies (eg, shortening) may be observed. The syndactyly may show fusion of the second and forth fingernails, which also may be seen in the toes. [10, 11] See the image below.
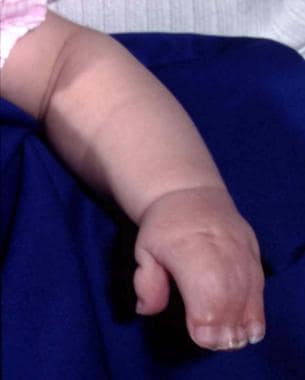 Typical Apert syndrome hand with syndactyly of all fingers. The thumb has been freed surgically.
Typical Apert syndrome hand with syndactyly of all fingers. The thumb has been freed surgically.
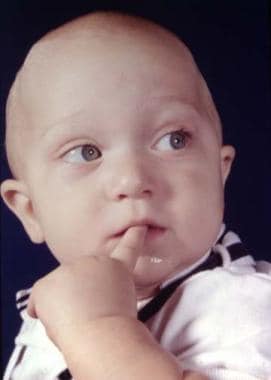
Upper eyelid ptosis with an antimongoloid slant may be seen. Blindness may be present. Overall, the deformity is worse than that of Crouzon syndrome. See the image below.
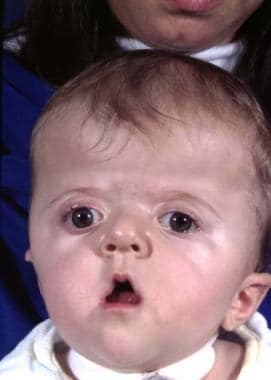 The classic features of Apert syndrome, including the broad skull, bulging in the temporal area, and retrusion and vertical shortening of the maxilla.
The classic features of Apert syndrome, including the broad skull, bulging in the temporal area, and retrusion and vertical shortening of the maxilla.
Pfeiffer Syndrome
This is an autosomal dominant condition caused by a single recurring mutation (Pro252Arg) of the FGFR1 gene and several mutations involving FGFR2. Patients have craniosynostosis, enlarged thumbs and great toes, and a hypoplastic midface. The hypoplastic midface gives the forehead an enlarged appearance. The nose is small. Exorbitism may be present, but it is never as prominent as in persons with Crouzon or Apert syndrome. The condition has been classified into 3 types. Patients with type I have the best long-term prognosis, whereas those with types II and III have neurologic compromise and die young. [12]
Saethre-Chotzen Syndrome
This is an autosomal dominant condition with full penetrance. It is caused by multiple mutations of FGFR2. Craniosynostosis is present, and the hairline is low. Ptosis and brachydactyly are characteristic. The forehead is retruded, giving the appearance of slight exorbitism. The maxilla may or may not be retruded.
Carpenter Syndrome
Patients with this autosomal recessive condition have craniosynostosis, syndactyly of the feet, and short hands and fingers with syndactyly of varying degrees.
Treatment of Craniosynostosis Syndromes
This collection of syndromes has proved to be an exciting area of investigation for plastic surgeons and other researchers. Surgeons currently have better tools to diagnose and investigate these syndromes, and they are now better understood. Most exciting of all, more sophisticated treatment methods have evolved over the past 20 years.
The age at presentation determines the treatment. If possible, provide early treatment and direct it at the cranial vault. The aim is to reduce raised intracranial pressure, if present, and to prevent visual problems. [13] In addition, the patient's appearance may be improved. The anterior cranial fossa enlargement is a result of frontal lobe growth; at 11 months, the frontal lobes are almost 50% of adult size. The anterior cranial base is at 56% of its total growth at birth and, at 2 years, has achieved 70% of its total growth, probably soon after this full growth is attained. The size and position of the anterior and middle cranial fossa floors are determined by the frontal and temporal lobes. If the anteroposterior growth of the skull base is diminished, this has no affect on mandibular growth.
Early skull base suture fusion results in interference with forward facial growth; thus, release of the affected sutures should provide normal growth. This certainly is the case for the skull, but the midface remains intruded in patients with Apert or Crouzon syndrome.
Undoubtedly, the best results are achieved in patients with isolated sagittal craniosynostosis. In the past, this was managed with a sagittal strip craniotomy. Currently, making coronal and lambdoid cuts to achieve immediate lateral expansion by hinging the cranial segments outwards is more common and is more effective in allowing the brain to expand. Do not interfere with the lambdoid and coronal sutures.
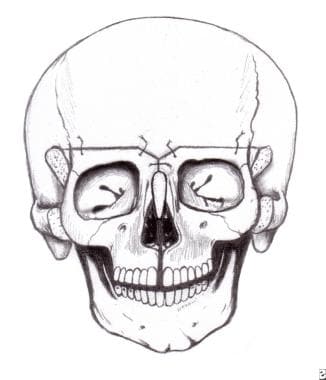
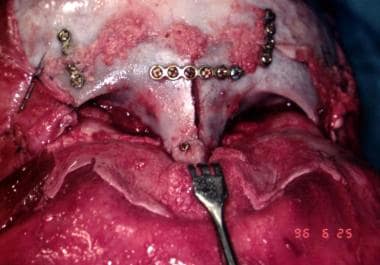
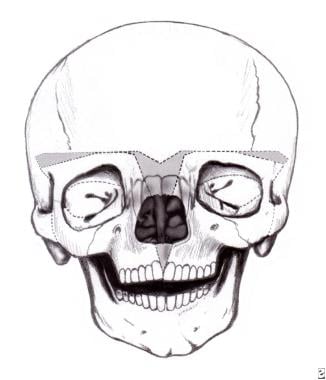
At 9-11 months in persons with bilateral coronal craniosynostosis, as is seen in Crouzon or Apert syndrome, the frontal area and supraorbital rims are osteotomized separately and advanced to produce a degree of overcorrection with improved frontal contour. [14] This also requires a cut through the orbital roofs and in front of the cribriform plate anteriorly. Laterally in the temporal fossa, the supraorbital rim is advanced using tongue-and-groove techniques for stabilization, although this is now not absolutely necessary because plates and screws (metal or preferably absorbable) are available. The frontal bone is now advanced and plated onto the newly positioned supraorbital rim. See the images below.
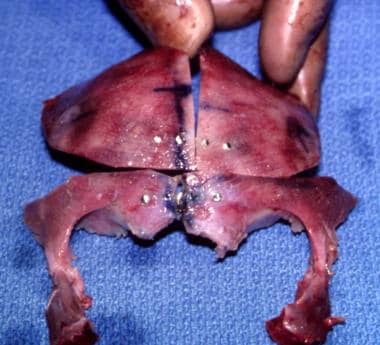 Surgical correction of Apert syndrome. The frontoorbital area has been removed and has been repositioned and remolded. It is ready to be inserted.
Surgical correction of Apert syndrome. The frontoorbital area has been removed and has been repositioned and remolded. It is ready to be inserted.
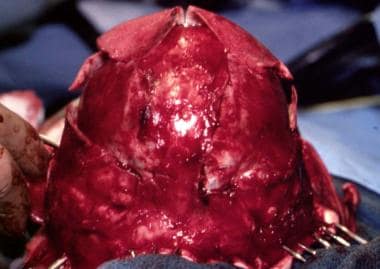 Surgical correction of Apert syndrome. The bony segment is back in its advanced position, and the scalp is ready to be closed.
Surgical correction of Apert syndrome. The bony segment is back in its advanced position, and the scalp is ready to be closed.
Even with large advancements, scalp closure is always possible. Occasionally, this is aided by galeal scoring. Although this is a standard technique, it is modified as required by the basic anatomy. The patient is monitored using postoperative CT scans to follow the resolution of the dead space. The placement of the coronal incision is very important. It should begin behind the ears and zig-zag as a large W-plasty across the occipital area. This avoids scars on the face and the visible part of the scalp. The zig-zag design allows the scar to be concealed with hair, even in bald patients who have a little hair on the posterior scalp.
Distraction
Currently, the trend is to advance and fix the frontosupraorbital region and to advance the maxilla subsequently or at the same time, even at an early age, with [15] or without [16] a distraction apparatus. Although internal distraction devices have been used to push the maxilla forward, with fixation on the temporal area and the use of a rod to apply an anterior pushing on the lateral orbital walls, these have not been uniformly successful. This is mainly because of design faults.
The rigid external distraction device, which is secured by skull fixation, with an anterior pulling force being applied to the maxilla, is more mechanically sound and works well. [17] It is expensive; the fixation can penetrate the cranium (should the patient fall, striking the side of the head), and the device can become displaced. [18] See the images below.
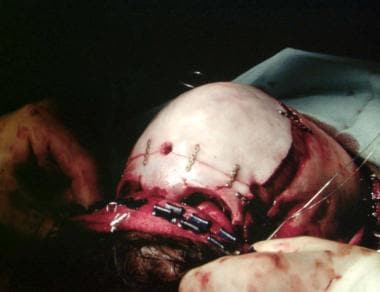 Osteotomies have been completed in the frontal and supraorbital areas. This complex has been advanced and fixed. Osteotomies have been performed on the maxilla, but it will be brought forward by external skeletal distraction.
Osteotomies have been completed in the frontal and supraorbital areas. This complex has been advanced and fixed. Osteotomies have been performed on the maxilla, but it will be brought forward by external skeletal distraction.
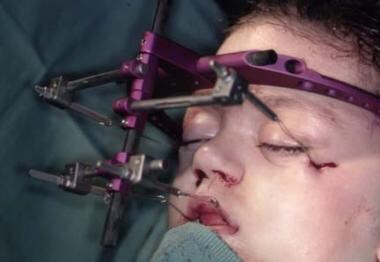
Despite this, good forward maxillary movement is obtained in a relatively controlled fashion. The children do not complain about wearing this device. It can be used for persons with Crouzon or Apert syndrome or other conditions that require an osteotomy and forward pull. [19] Whether this is an advantage over the older, well-established, and safe method of advancement osteotomy, stabilization, and bone grafting in one stage remains unproved. In older patients, the latter method probably remains the technique of choice.
A study by Hammoudeh et al indicated that simultaneous Le Fort III/I surgery (midface advancement and orthognathic surgery) is a relatively safe treatment approach. Major and minor complications were experienced by 12% and 40% of patients, respectively, with Le Fort III and I advancements averaging 6.18 mm and 6.70 mm, respectively. The investigators advised that select patients with global midface retrusion and class III malocclusion be considered for such surgery. [20]
Pierre Robin Sequence
This condition consists of micrognathia, glossoptosis, and, frequently, a cleft of the palate. Airway obstruction occurs in the more severe cases. The mandible is retrognathic, and the genioglossus muscles are shortened; thus, the tongue falls back and occludes the airway. The degree of this latter anatomic anomaly varies, but it is always present and can be lethal.
The deformity may be associated with the position of the baby in the uterus, with its head flexed for a longer period than normal. In some patients, mandibular growth after birth is good, in contrast to those who remain significantly micrognathic. The child has a birdlike face with varying degrees of retrognathia and respiratory problems. He or she also may have feeding difficulties. Respiratory obstruction is present because the tongue falls back into the pharynx. The intercostal spaces retract, and the accessory muscles of respiration are overactive. The baby tends to choke, and cyanosis can be ever-present or intermittent. This occurs especially during sleep.
Feeding difficulties
These are caused by a combination of the small mandible, the position of the tongue, respiratory distress, and cleft palate. The child only takes small amounts, coughs frequently because food is inhaled, and regurgitates much of the food; tracheostomy and gavage feeding may be necessary. Associated with this are malnutrition, failure to gain weight, frequent chest infections, and exhaustion. In the presence of a complete cleft, the tongue may cause respiratory problems by going through the cleft and into the nose. The large appearance of the tongue is probably relative; the mandible is small. Other airway problems should be sought, including tracheoesophageal fistula and choanal atresia. The child should be checked for other conditions that can be associated, such as cardiac, limb, and eye problems.
Treatment
The condition can range from minor to severe; therefore, treatment varies. [21, 22, 23, 24, 25, 26] See the images below.
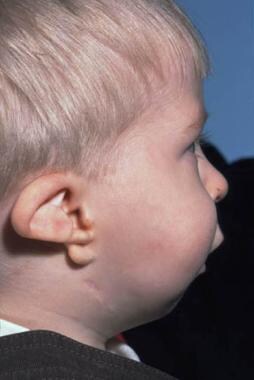
Mild cases are treated by placing the baby on its side or face down with the foot of the cot raised. This allows the tongue and jaw to fall into a better position and may be totally adequate. This placement allows the child to sleep, which is vital. The condition improves as the child becomes bigger and stronger.
If this maneuver is unsuccessful, the next step is intubation. This can be difficult and should be performed with the child awake. The tongue can also be advanced using a suture passed through it. It can be sutured to the lip mucosa or a Kirschner wire can be passed through its base. In this situation, the child must be watched carefully by a trained staff member. An oxygen monitor should be placed in addition to a cardiac monitor.
If the child continues to need an endotracheal tube after 2-3 days or cannot control its airway after a week of conservative treatment, consider surgery. A tracheostomy can be difficult in these small children; if possible, a Björk flap should be used. This prevents the tracheostomy from closing, allows for greater ease of changing endotracheal tubes, and maintains patency of the tracheostome. [27]
Suturing the tongue to the lower lip to open the airway has become more popular. This can deform the lip and tongue, but it may be effective for opening the airway. Other procedures involve fixation of the tongue in a forward position using a Kirschner wire and many other variations of the tongue-to-lip adhesion.
The introduction of mandibular distraction has yielded the possibility of immediate correction of the deformity by lengthening the mandibular body bilaterally. [28] The distractor is applied to the body of the mandible, and a partial body osteotomy with preservation of the mandibular nerve is performed. The distractor may be external or internal; the latter is almost becoming standard in the author's practice. It is easy to perform and is simple for the parents to manage. The procedure is performed bilaterally, and the mandibular body is lengthened; thus, the tongue is brought forward. A period of consolidation occurs once a slight overcorrection has been achieved. The external method is used in many centers and is equally effective. This is a logical method that corrects the primary deformity. The other methods have been less effective and have resulted in deformity.
Predicting how these children will fare in the long term is always difficult. Some develop normally, and others may require further mandibular surgery to achieve an acceptable facial profile. Proper care by experienced surgeons in good centers has greatly improved the outlook for these children. Most should survive if the proper decisions are made at the correct times. Management by experienced surgeons, anesthesiologists, and nurses is essential
Hemifacial Deformity
Hemicraniofacial Microsomia
Poswillo has suggested that this condition results from bleeding in the temporal area in the embryo. [29] It has also been associated with thalidomide intake during pregnancy. If epibulbar dermoids and vertebral anomalies are present, it is termed Goldenhar syndrome. This condition can be bilateral. The components of the syndrome are low-set ears, short mandibular ramus, micrognathia, chin deviation, occlusal cant, and anterior open bite. See the images below.

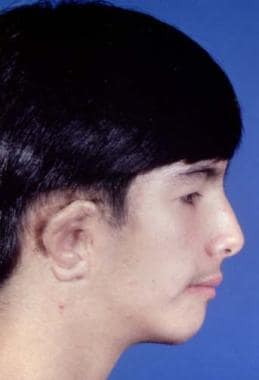
The zygomatic arch may be present, absent, hypoplastic, or partially absent. Severe deficiency of the subcutaneous soft tissue and absence of temporal hair may be observed. The orbit may be displaced, deformed, and small. The eyes may be rudimentary or absent, and the lateral canthus can be displaced. The side of the face may be hypoplastic or vertically short. All degrees and variations of this deformity may occur. Many classifications schemes have been developed, but most are unhelpful.
A careful assessment of both the hard and soft tissue deficiencies is advised, while keeping treatment considerations foremost in mind. The major features can be considered as auricular, maxillary, and mandibular underdevelopment. In severe cases, the skull is also involved, especially the sphenoid and temporal bones.
The mandibular deformity can range from (1) minimal hypoplasia to (2) a small ascending ramus and condyle with an absent glenoid fossa and, possibly an absent coronoid to (3) a minimal or absent ramus with severe vertical facial shortening.
These deformities become worse with time. Flattening and hypoplasia of the zygomas and its arch (the latter may be absent) also may occur. The muscles of mastication (ie, masseter, medial and lateral pterygoids, temporalis) may be hypoplastic. This upsets the functional matrix, which further affects related bony development.
Ear deformities vary in severity. Grade 1 is small, malformed ears. Grade 2 is a vertical remnant of skin and cartilage with no ear canal. Grade 3 is a small remnant and lobule. Patients also have variable degrees of deafness. Audiometry and CT scanning are imperative. Look for possible cerebral anomalies.
Cranial nerves
Abnormalities are common in patients with craniofacial microsomia. The facial nerve is most commonly affected to a greater or lesser degree. Optic, trigeminal, cochlear, and cranial nerves are involved.
Facial clefts
The distance between the eye and the angle of the mouth is shortened. Transverse facial clefts are seen and vary in severity. They span the angle of the mouth to the ear.
Renal
The patient may have a hypoplastic or missing kidney.
Investigations
Panorex, cephalograms, CT scans, 3-dimensional CT scans, and MRI are all helpful in the assessment of the deformity. Dental impressions are taken.
Treatment
Treatment is long-term. Unilateral or bilateral mandibular distraction can be performed early, especially in patients with bilateral deformity and airway problems. See the images below.
 Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
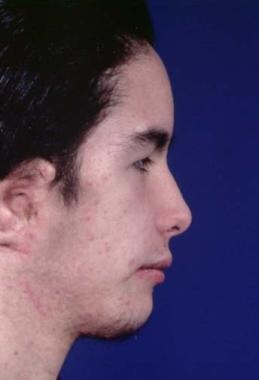 Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
The skull deformity and orbital deformity can be corrected before the child starts school. Skull grafts can be used to reconstruct defects in the orbit, zygomatic arch, and malar area. When the ascending ramus of the mandible requires reconstruction, a costochondral graft can be effective. Later, a Le Fort I and sagittal split mandibular osteotomy is performed with chin advancement as necessary. When an ascending ramus is required, a rib graft can be effective. Soft tissue is supplied by free tissue transfer.
The facial cleft is repaired in layers. As in the cleft lip, the muscle layer, frequently disregarded in the past, is most important and must be repaired accurately.
Orthodontic supervision is essential, and further maxillary and mandibular osteotomies are frequently necessary when the face is mature (age >16 y).
Even with all these well-planned procedures performed as indicated over the years, the end result may leave much to be desired.
Romberg Disease (Progressive Hemifacial Atrophy)
Romberg disease, also known as Parry-Romberg syndrome, is a sporadic condition in which progressive atrophy of skin, subcutaneous fat, muscle, and bone occurs. [30] The contralateral face may be involved, but this is uncommon. The process can go on for a variable number of years. The skin and adnexa atrophy, fat becomes inflamed, and muscle atrophies. Nerves are unaffected. An association with sympathetic stimulation has been postulated, but the cause remains unknown. See the image below.
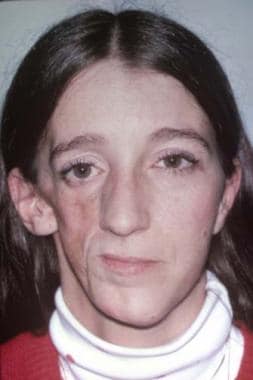 Preoperative appearance showing atrophy of the right side of the face involving both soft tissue and bone.
Preoperative appearance showing atrophy of the right side of the face involving both soft tissue and bone.
A report by De la Garza-Ramos et al found that 48 out of 80 study patients with Romberg disease (60%) had brain abnormalities, as seen on magnetic resonance imaging (MRI) scans, with these abnormalities being ipsilateral to the hemifacial atrophy in 26 of the 48 individuals (54%). The predominant brain finding, white matter disease, was more frequent and severe in those individuals with epilepsy, as was hemispheric atrophy. [31]
Differential diagnosis
Lipodystrophy produces a similar appearance, but only in fat and is often bilateral. Scleroderma is similar to hemifacial atrophy.
Treatment
Treatment should be delayed until the process has ended.
Bone
This can be augmented by iliac crest or split skull, the latter being taken from the contralateral side. Fixation should be rigid. The skull can be also augmented with methyl methacrylate or hydroxyapatite cement (Bone Source, Stryker Leibinger, Freiburg, Germany; Mimix, Biomet, Warsaw, Ind; or Norian CRS, Synthes-Stratec, Oberdorf, Switzerland).
Soft tissue
This should be replaced via transfer of vascularized free tissue, deepithelialized skin, subcutaneous tissue, and fat. See the image below.
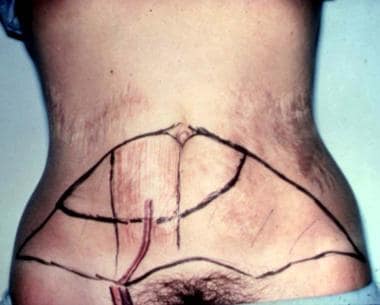
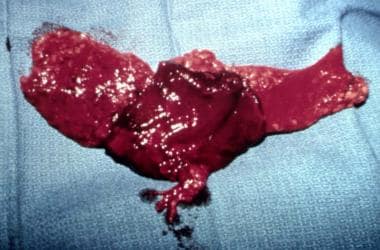
Muscle can be incorporated. The use of muscle alone is not recommended because it tends to atrophy over time. Occasionally, a skin island is used to release and augment the skin deficiency. The free tissue transfer should be securely fixed, preferably to bone with nonabsorbable sutures to avoid slippage. Omentum can be used, but it tends to sag even with secure and plentiful fixation from a vascular standpoint. Pedicled galea is available but is limited to cheek defects of small volume and is not always reliable. Tongue flaps can provide filling material for atrophic lips. Dermis can also be used, but this frequently loses considerable volume. Rarely is total symmetry achieved. See the image below.
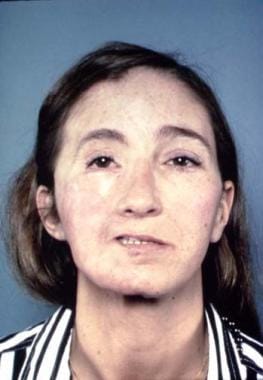 End result following reconstruction of right side of face with rectus myocutaneous free flap and bone grafting.
End result following reconstruction of right side of face with rectus myocutaneous free flap and bone grafting.
Klippel-Feil and Turner Syndromes
Klippel-Feil Syndrome
This condition has 3 radiologic types. The first is characterized by continuity of cervical and upper thoracic vertebrae. The second is characterized by involvement of only a few interspaces but with hemivertebrae with scoliosis and fusion of the atlas to the skull base. The third is characterized by fusion of the cervical, lower thoracic, and lumbar vertebrae. [32, 33]
Clinical features
Patients have a short neck, restriction of neck movement (especially rotation), and a low occipital hairline. Flexion and extension is better than rotation. Associated features are webbing of the neck, scoliosis, torticollis, and, occasionally, Sprengel deformity, which is displacement of one or both scapulae. In some cases, patients have intellectual disability. The patient also may have problems with eye movement, hearing loss, and urinary anomalies. One third of patients have unilateral renal agenesis, and vaginal agenesis has also been noted in a few children.
The syndrome occurs in 1 in 42,000 births and is more frequent in females than males. Type I is most common clinically, and type II causes fewer clinical problems and is often found only at autopsy. Palatal clefts alone tend to occur 3 times more frequently than combined lip and palate clefts. Cardiac anomalies, especially ventricular septal defects, are more common in affected children than in healthy children.
Other syndromes with similar cervical findings are Noonan and Morquio syndromes; spinal tuberculosis can have a similar involvement. The cause is unknown.
Cause
The process is failure of segmentation of the cervical spine. The cause of this has been postulated to be a lack of midline fusion with disruption of the notochord. A second theory is that the subclavian artery has been disrupted.
Treatment
This depends on the extent of the problem, which may consist of cardiac, palatal, and skeletal involvement. If cervical stability is in question, anesthesia may be a problem. It must be performed with care to prevent any neurologic problems. Spinal fusion should be performed, and cord pressure should be decompressed. Correction of neck webbing is difficult and consists of resection of excess skin and underlying soft tissue with Z-plasty skin closure. The platysma rather than the deep muscles are involved. Other types of skin plasties have been described, but all produce unsatisfactory scars. The posterior hairline is corrected by expansion of the neck skin with subsequent excision of the hair-bearing area with expanded skin advancement.
Turner Syndrome
This occurs in females. A webbed neck, infantilism, and cubitus valgus are observed. Another term for this condition is pterygium colli. Patients have ovarian agenesis and dwarfism. These girls have only 44 autosomes and one sex chromosome, the X-O.
The findings are dwarfism, webbed neck, low hairline, epicanthal folds, mandibular deformity, webbed elbows and knees, coarctation of the aorta, hypertension, lymphedema of the hands and feet, and intellectual disability. A buccal smear is taken, and the absence of a peripheral chromatin mass confirms the diagnosis.
Treatment
A gynecologist should treat these patients in conjunction with an endocrinologist. Webbing of the neck can be corrected with an unequal Z-plasty. Hair is excised from the lateral hair-bearing area, and a small hair-bearing flap is transposed into a lateral defect resulting from a large flap being taken and placed posteriorly to raise the posterior hairline. Another method, as mentioned earlier, is to expand non–hair-bearing skin and advance this after excision of the posterior hairline. If only the lateral web needs to be addressed, multiple Z-plasties may be used; however, the scars can be obvious and may be of poor quality.
Various midline resections can be made on the back of the neck, with advancement of the skin edges in different designs, such as advancement lateral to medial with X or Y scars. Although improvement of the webbing is achieved, it is never complete and the scars (eg, keloid, hypertrophic, stretched) can be most unsatisfactory.
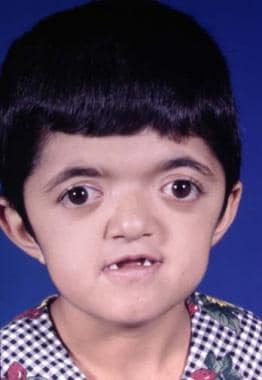
Treacher Collins Syndrome (Mandibulofacial Craniosynostosis)
This condition is passed on as an autosomal dominant gene with variable penetrance and phenotypic expression. It is a bilateral condition that affects first and second arch structures. As with all inherited conditions, it varies from mild to severe. Patients have hypoplasia of the zygomas and mandibles with ear defects or an absence and displacement of the sideburns. The eye area can be significantly involved, with an antimongoloid slant of the fissures, colobomas of the lower lids, and an absence of eyelashes medially. [34] See the images below.
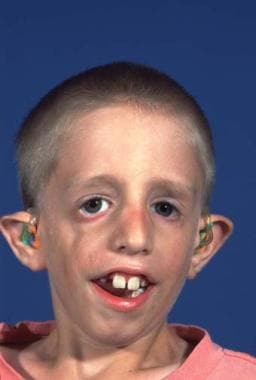
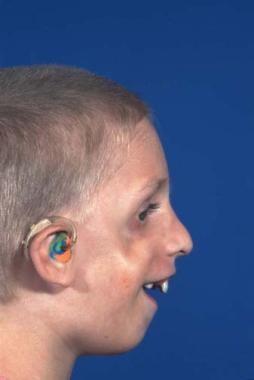
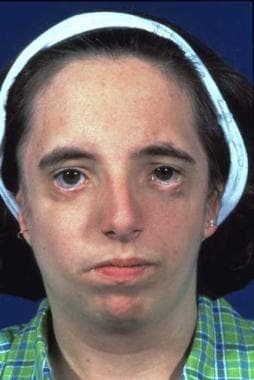
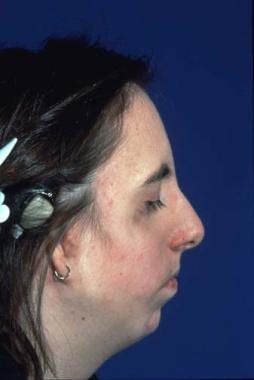
The nose is beaked downwards at its tip. In severe cases, patients have thinning of the subcutaneous tissue from the angles of the mouth up to the lateral canthus. This is associated with macrostomia and is similar to a subcutaneous cleft. The texture of the skin in this area is different because of its thinness. It is also known as Franceschetti-Klein syndrome. The face is fishlike, and patients may have deafness and intellectual disability.
Other associated problems include vertebral anomalies, clubfoot, lung agenesis, and frontalis agenesis. The hypoplastic mandible, glossoptosis, small size of the pharynx and nasopharynx, and occasionally choanal atresia can cause severe breathing problems and even death. A more minor situation causes sleep apnea, with all of its attendant problems.
Patients have a family history positive for the syndrome. The condition is transmitted by autosomal dominance, and the penetrance and expressivity are variable. The mother transmits the gene, and it may become more lethal from generation to generation. The syndrome seems to be more prevalent in children born to parents of older age. An increased incidence of spontaneous abortions is observed.
Differential diagnoses include Nager syndrome and ablepharon macrostomia syndrome. The syndrome may be due to problems of differentiation of the branchial arch mesoderm interfering with facial bone development. Stapedial artery hypoplasia may cause ischemia in the facial region. A significant portion of the craniofacial area, soft tissue and cartilage, bone, and teeth are derived from the neural crest ectoderm. The gene for Treacher Collins syndrome has been mapped to band 5q31.3-q33.3. [35] This has been reproduced in an animal model by damaging neural crest cells using vitamin A.
Overall management
These patients require treatment by a multidisciplinary team, including a plastic surgeon, oral surgeon, orthodontist, oculoplastic surgeon, and, occasionally, a neurosurgeon. The social worker plays an important part in the social aspect of the family.
Shortly after birth, the patient may develop airway problems; the help of an otolaryngologist may be necessary. In some cases, simple head-down positioning with elevation of the foot of the crib may be sufficient; in others, a tracheostomy is considered. More recently, as described for Pierre Robin sequence, early bilateral mandibular distraction can be performed to correct the airway problem.
Others involved with the treatment of these children are audiologists and speech pathologists. Later, they may require a psychiatrist. Feeding difficulties may require the help of a nutritionist.
Skeletal assessment
This is essential prior to contemplating treatment. Use standard facial bone radiographs, CT scans, and 3-dimensional CT scans.
The orbital shape shows an inferolateral rotation with an absence of the floor. The malar area and zygomatic arch are missing to a variable degree. The central portion of the maxilla seems somewhat protruded and vertically shortened; it is also narrower than normal. The mandible shows an increasing deformity from hypoplasia of the glenoid fossa and ascending ramus to complete absence of these areas with a free-floating mandible. The classifications are type I, type II subtypes A and B, and type III. The facial soft tissue is also disrupted.
Treatment
The mandible can be treated early using bilateral osteotomies in the body with distraction, either intraorally or extraorally. Many different methods have been described for the orbitomaxillary reconstruction. The requirements are a recontour of the orbit and a reconstruction of the lateral orbital wall and orbital floor together with the zygomatic arch and malar region.
Many types of bone have been used in the reconstruction, including rib, split skull, vascularized cranial bone on temporalis muscle, and iliac crest bone grafts. The author has tried all of these and has developed a reconstruction algorithm.
At approximately age 5-6 years or when the patient presents to the surgeon, the problem is assessed using clinical examination and 3-dimensional CT scans. Minor deformities are augmented with cranial or iliac crest bone grafts rigidly fixed with screws and, if necessary, plates. This can be performed from an intraoral approach. With more significant bone absence, a coronal flap exposure is used to expose the orbital and malar area. The orbital contents are radically freed from the facial soft tissues and any bony attachments so that they can be reduced into the orbit. A model of the missing bone, zygomatic arch, and malar is obtained. This is then transferred to the temporal area, and the design is marked on the skull with a pencil.
A full-thickness cranial bone graft is taken by cutting along the planned osteotomies using a contouring burr or an air drill. A microplate is then contoured to form the shape of the zygomatic arch and the lateral aspect of the malar area. See the images below.
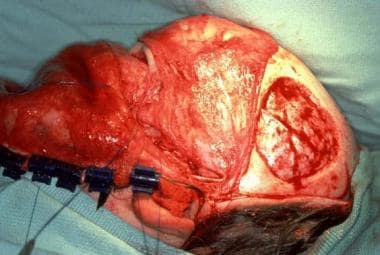 Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
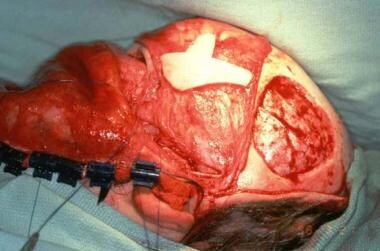 Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
The bone graft is osteotomized to fit onto the shaped microplate. The latter is then screwed onto the malar bone and the small nubbin of bone in the preauricular area, which represents the rudimentary lateral origin of the arch. [36] See the images below.
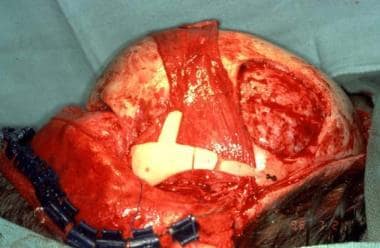 Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
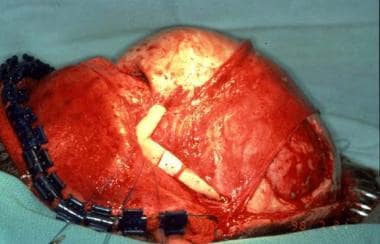 Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
Further skull bone grafts are taken as required for the lateral orbital rim and the lateral aspect of the supraorbital rim. Prior to this, the junction of the supraorbital and lateral orbital rim is contoured to a normal shape. The bone grafts are fixed with screws. In more severe cases, a significant defect remains in the floor, particularly in the lateral area. This is also reconstructed with a bone graft, which often must be wired into position. In patients with larger lateral defects, stacked grafts are used.
Attention is now turned to the skull defect. This is filled with bone shavings taken from the skull with a sharp osteotome. From the posterior scalp, a large sheet of pericranium is raised. This is used to cover the zygomatic arch reconstruction and to smooth out the whole area. Fortunately, the upper lids have an excess of skin. After closure of the scalp, attention is turned to the eyelids. The excess of skin is taken down as a flap based laterally. The skin of the tight lateral area of the lower lid is totally released, and the upper lid flap is inserted into the defect. This allows the lateral canthus to be elevated and fixed to the inner aspect of the lateral orbital rim. Overcorrection is advised.
This procedure corrects much of the deformity; however, a residual vertical shortage of the lower lid remains laterally. This occurs because of a deficiency of the internal lamella of the lid. After 3-6 months, this is corrected. Recently, a newly described method of internal lamella reconstruction has been described that has, so far, yielded the best result. The lid is injected with a vasoconstrictor and is released on its inner aspect using a horizontal incision.
When the correct height is obtained, a shaped conchal cartilage graft, with intact perichondrium on the surface facing the eye, is sutured into place with buried sutures. The perichondrium epithelializes, and a good result is usually achieved. In the past, a full-thickness upper lid island flap was used, but the present method has been found to be more suitable. If lateral canthal adjustment is required, this can be performed concurrently.
The mandibular retrusion can now be managed using distraction osteogenesis. [37] Complex cases require glenoid fossa and perhaps ascending ramus reconstruction. Costochondral grafts are occasionally required for ascending ramus reconstruction. The author has found that temporal skull augmentation is frequently required later. This is performed using alloplastic material, methyl methacrylate, or one of the hydroxyapatite pastes. From the unfortunate complications described by the authors' team and others, the hydroxyapatite paste reconstruction is not now used. [38, 39]
A study by Ali-Khan et al indicated that in patients with Treacher Collins syndrome who undergo bilateral mandibular distraction osteogenesis, airway function outcomes are poorer than in patients with Pierre Robin sequence who are treated with the procedure. Surgery in the study was deemed successful if imminent tracheostomy was prevented or the patient was successfully decannulated within 1 year subsequent to primary distraction. Such success was achieved in 21% of patients with Treacher Collins syndrome and 65% of patients with Pierre Robin sequence, with repeated distraction required in 46% and 7.7% of patients, respectively. [40]
Later reconstruction
In adolescence, after orthodontic management, a bimaxillary procedure is performed. Usually, this consists of a Le Fort I and a sagittal split advancement of the mandible with an advancement genioplasty.
The author typically performs the necessary upper face augmentation using a coronal flap approach. Skull bone grafts are favored for the reconstruction. At the same time, a rhinoplasty with any necessary airway surgery is performed. In some cases, nasal augmentation is required. This is accomplished with a split- or full-thickness cranial bone graft, depending on the required dorsal shape. Occasionally, a repeat lateral canthopexy is required. In all of these cases, remember that the problem with the airway remains regardless of the patient's appearance; consequently, all of the precautions necessary in such a situation must be observed.
These patients have greatly improved appearance; thus, their confidence increases. See the images below.

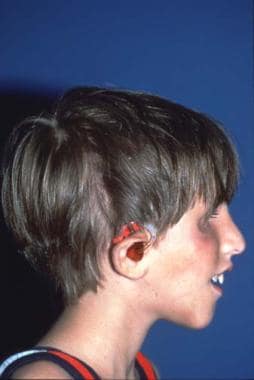
 Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
 Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
The beneficial results of the surgery bring both increased job opportunities and better chances to meet others, which leads to a more normal social life. One problem is that the Treacher Collins condition may become more common as more people with the condition have children and pass the trait to their offspring.
The ear reconstruction is performed at age 6 years using costal cartilage in the manner described by Brent and others. [41]
Hypertelorism and Hypotelorism
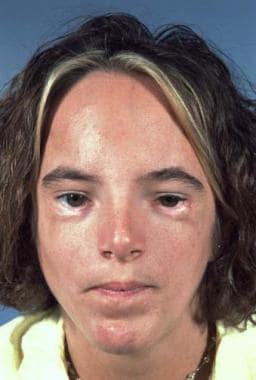
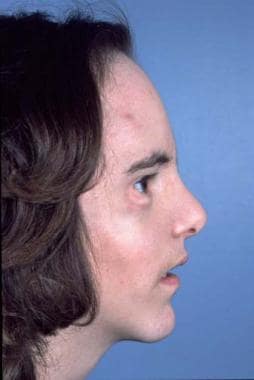
Hypertelorism (Greig Syndrome)
In persons with this condition, the orbits and their contents are shifted laterally, either by a midline cleft or excessive ethmoid sinuses. [42, 43] The anterior skull may be flat, and the frontal area may be somewhat retruded. The maxilla may be hypoplastic and/or retruded with a class III dental occlusion. Often, diastasis of the upper incisor teeth is seen. The nose may be bifid. In rare cases, the midface and palate may be widely clefted. The condition may be asymmetric, but this is usually associated with clefts. The patient's level of intelligence may vary from normal to significantly reduced. See the images below.
 Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
 Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
Treatment
Minor degrees of deformity (referred to as telecanthus) can be corrected by removing a small amount of bone in the midline. [44, 45, 46]
Osteotomies of the medial orbital walls are performed, and the walls are moved together and stabilized to the glabellar areas and to one another with wires or miniplates. The approach is by a coronal flap. Usually, one need not remove skin; however, an ellipse can be resected if indicated. In some cases, the nose is flat and may require reconstruction with a cranial bone graft. When the intercanthal distance is such that a conservative approach will not yield a good result and when a degree of downward rotation of the orbit is present laterally, a more involved operation is necessary. A coronal approach is used to explore the frontoorbital region. The temporalis muscles are dissected from the fossa.
A limited frontal craniotomy is performed in the midline. This allows a total orbital osteotomy to be performed. This is carried out after the central bony block of a precalculated width is removed. The central cribriform area is removed together with the frontal sinuses. The orbits are mobilized and brought together. If necessary, the lateral orbits are rotated superiorly. This allows correction of the antimongoloid slant by superior fixation of the lateral canthal ligaments. A cranial bone graft may be necessary for correction of the flat nasal bridge line. See the images below.
 Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
 Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
When the hypertelorism is wide with a midface cleft or when the dental arch is distorted because of vertical maxillary shortening in the midline, a different and more satisfactory procedure termed the facial bipartition is used. [45, 46] See the images below.
 Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
 Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
Through a coronal approach, a V-shaped osteotomy is formed from the frontal area down to the nasal bones. With a fine osteotome, an osteotomy is made between the upper incisions and the cut is continued along the hard palate. The zygomatic arch is cut, the retrotuberosity osteotomy is performed, and all walls of the orbit are osteotomized. See the images below.
An osteotomy is made on the orbital roofs to meet the medial and lateral wall osteotomies. With maxillary mobilizing forceps, the 2 segments are loosened. Now they can be rotated together superiorly using the incisor osteotomy as the center of the arc of rotation. The orbits are brought together in their correct axis in 3 dimensions. This is a stable osteotomy after it is fixed. The soft tissue is reapplied, placing the lateral canthi in their correct positions. The nose may require bone grafting. If the patient has a midline facial cleft, it is repaired in layers. All bony fixation is with plates and screws. See the images below.
Hypotelorism
This is a rare condition that may not be compatible with life. In the most severe cases, the infant looks like a cyclops. The anatomy is complex. In most cases, the intercanthal area is narrowed, and varying degrees of frontonasal hypoplasia are seen. The orbits are large and may be oddly shaped. Frequently, the patient has a lower-than-normal level of intelligence.
Treatment
See also Trigonocephaly - Metopic craniosynostosis. The frontonasal supraorbital area is exposed through a coronal approach. Bilateral orbital osteotomies are performed, and the orbits are moved apart. The central defect is grafted with bone. Usually, a bone graft is used to supply a nasal bridge line. Characteristically, the nose is long and cannot be effectively shortened. The medial canthi are repositioned. When this has been achieved, a soft tissue defect is formed, which can be reconstructed using a local flap. The end result can be satisfactory, but, undoubtedly, the central portion of the face is long. Associated upper and/or lower lid colobomas may be seen. These must be repaired in layers in a standard fashion. The best plan is to correct these prior to performing the orbital repositioning.
Craniosynostosis
History
This condition has been known since antiquity. Hippocrates was the first to describe it in 100 BC. Others reported deformed skulls without sutures. Even in these early days, premature closure of the sutures was known to cause specific skull deformities. Under this heading, isolated craniosynostoses are compared to syndromes of craniofacial synostoses such as Apert and Crouzon syndromes, which are considered separately.
Common deformities that occur as a result of premature suture fusion are scaphocephaly, trigonocephaly, brachycephaly, plagiocephaly, oxycephaly, and turricephaly. The sutures involved in these deformities are sagittal, metopic, bicoronal, unicoronal, and multiple for the last 2 conditions. Complex premature suture fusion is associated with Apert, Crouzon, Kleeblattschädel, Pfeiffer, Saethre-Chotzen, and Carpenter syndromes. The cause of the premature fusion remains unknown.
Symptoms
In many cases of craniosynostosis, patients have no true symptoms, only deformity. Single-suture craniosynostosis rarely produces a significant rise in intracranial pressure, but 2 or more can certainly cause this. [47, 48] When the increased pressure is established, the brain pushes against the skull to cause "thumb printing" or, in some cases, true erosion over the gyri. This is a definite indication for surgery. Hydrocephalus may be present and should be assessed with the placement of a ventriculoperitoneal shunt if the degree of pressure warrants such a measure. [49] A further complication of this situation is blindness. [50]
Visual deterioration is associated with elevated intracranial pressure. This is usually observed in multiple-suture stenoses (eg, oxycephaly, brachycephaly, Crouzon syndrome, Apert syndrome). It is rarely observed in plagiocephaly, scaphocephaly, and trigonocephaly. The cause of blindness is not actually known because the optic canals are of normal dimensions. It may result from increased intracranial pressure, a change in the skull base dimensions, or chronic papilledema. In patients with severe hypertelorism, their wide-set eyes may prevent them from developing binocular vision.
Craniofacial Development
Knowledge of the time scale of craniofacial growth is important. The anterior cranial fossa enlarges as the frontal lobes enlarge with growth. They are 47% of adult size at 11 months and 93% of adult size at age 7 years. By age 2 years, the anterior portion of the cranial base is at 70% of its total growth. The situation with the temporal lobes is somewhat similar. The nasomaxillary complex follows the growth of the anterior skull base.
Plagiocephaly - Unilateral Coronal Craniosynostosis
When one coronal suture fuses prematurely, the frontal area is flat, the supraorbital rim and the lateral orbital wall are retruded, and the orbit is narrowed transversely. Coronal synostosis is due to an amino acid substitution (Pro250Arg) that results from a single-point mutation in the FGFR3 gene on arm 4p. [51] See the image below.

The palpebral fissure is narrowed, and the eyebrows are elevated laterally. The nose may be deviated to the affected side, and the ear may be displaced anteriorly. Radiologically, the orbit shows the harlequin deformity due to the elevation and retrodisplacement of the lesser wing of the sphenoid. [52] Frequently, occipital bulging is observed. The condition is rare, occurring in 1 in 10,000 live births. [53] This situation should not be confused with deformational unilateral skull deformity.
Treatment
Correction is performed when the patient is aged 6 months, if possible. [54, 55] Several methods are available for treating this deformity. The involved portion of frontal bone is removed, and this may be rearranged by osteotomies or by barrel-staving. In the latter, a series of parallel cuts are made through the bone until it can be reshaped. See the images below.
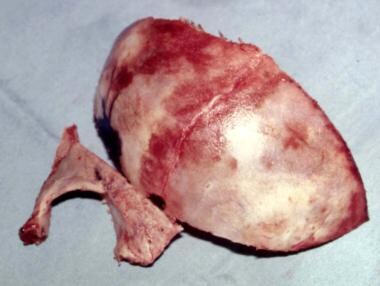 Unilateral coronal craniosynostosis. The frontal bone and the right supraorbital and lateral orbital wall have been removed.
Unilateral coronal craniosynostosis. The frontal bone and the right supraorbital and lateral orbital wall have been removed.
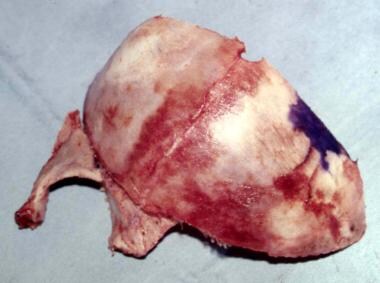 Unilateral coronal craniosynostosis. The skull is rotated so that the flat part lies posteriorly on the opposite side.
Unilateral coronal craniosynostosis. The skull is rotated so that the flat part lies posteriorly on the opposite side.
The lateral orbital wall and supraorbital rim are modified by osteotomies. See the image below.
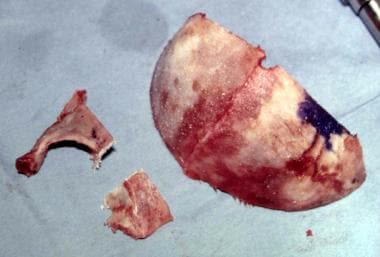
When the desired shape is achieved by whatever method is chosen, this complex is replaced and stabilized by resorbable plates made from polylactate combined with polyglycolate. Titanium microplates can be used to assemble bone plates, but these are then placed onto inner-aspect segments to obviate any erosion towards the brain and to eliminate any irregularities under the scalp. See the images below.
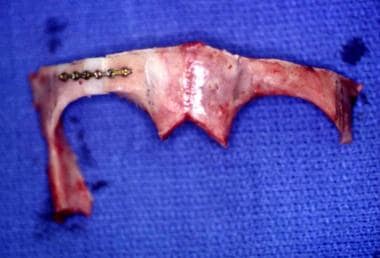 Unilateral coronal craniosynostosis. Bone graft is inserted to make the right orbit the same size as the left orbit.
Unilateral coronal craniosynostosis. Bone graft is inserted to make the right orbit the same size as the left orbit.
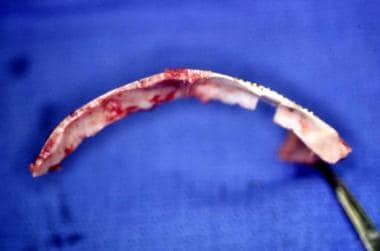
 Frequently, the supraorbital rim has to be rotated anteriorly as can be seen from this view.
Frequently, the supraorbital rim has to be rotated anteriorly as can be seen from this view.
In an alternative method, the supraorbital rim is expanded by inserting a small cranial bone graft into a central osteotomy. The size of the graft is determined based on measurements of the normal supraorbital rim. The frontal area is osteotomized and rearranged as necessary. Again, plates (absorbable or metallic) are used on the intracranial surface and to fix the cranium and supraorbital rim together. Absorbable plates secure the skull and supraorbital segment in place, if the whole misshapen orbito-orbitofrontal segment is totally remodeled, to provide symmetry with arrangement; reconstruction and fixation are performed rapidly and securely. See the image below.

Deformational Unilateral Skull Deformity
The sutures are intact. However, because of the child's position in the uterus, the head is asymmetric but the orbit is less involved, no harlequin sign develops, no nasal deviation occurs to the ipsilateral side, and no ridge forms over the coronal suture. Other causes include the practice of placing sleeping infants on their backs in order to reduce the risk of sudden infant death syndrome [56] and a greater number of infants being referred for early assessment. [57]
It occurs in 5-25% of babies. [58, 59] Some degree of occipital flattening occurs. When the skull is viewed from above, the ear is markedly displaced backwards on the flattened side. Usually, this condition is self-correcting; however, occasionally, surgical correction is indicated. [60] Helmet treatment can also be used, but ideally, this should occur within the first 6 months of life. [61]
Differentiation
On CT scan images, especially 3-dimensional CT scan images, the lambdoid suture is open. Posterior flattening occurs along with contralateral anterior flattening. The ear on the affected side is displaced anteriorly when viewed from above.
Treatment
Surgical treatment is necessary for unilateral craniosynostosis or severe postural deformity. If the orbit is constricted, it is enlarged by a vertical osteotomy in the supraorbital rim and a bone graft is placed. The coronal osteotomy is rotated to place the flattened area under the hair-bearing scalp on the noninvolved side. A lateral canthopexy is required, and, rarely, a medial canthopexy must be performed. [54] Consider helmet treatment in less severe cases. [61]
Bilateral Coronal Craniosynostosis
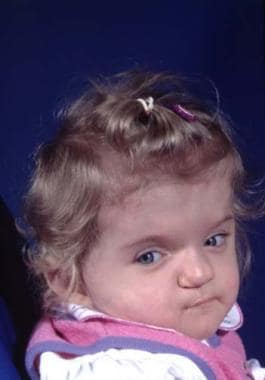
Both coronal sutures are fused, resulting in a widened head with a decrease in anteroposterior dimensions. The supraorbital rim is recessed, and the eyes are prominent. The face is usually unaffected. See the image below.
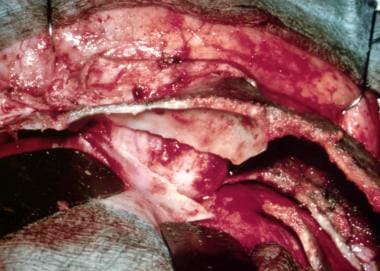
This condition can be nonsyndromic or syndromic when a component of Crouzon, Apert, Jackson-Weiss, or Pfeiffer syndrome is present. This causes variable degrees of brachycephaly. The syndromes can be defined at a molecular level. [62] Pfeiffer syndrome is heterogenous with a single recurrent mutation (Pro252Arg) of the FGFR1 gene and mutations affecting FGFR2. Crouzon syndrome is associated with multiple mutations of FGFR2. Jackson-Weiss syndrome is associated with an FGFR2 mutation. Apert syndrome is associated with an FGFR2 mutation.
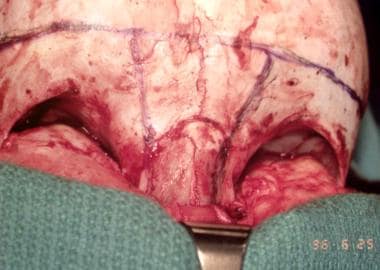
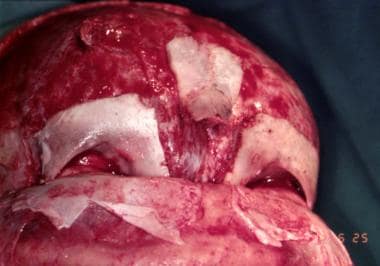
Surgery is performed when the patient is aged approximately 6 months, but it can be performed earlier. Treatment begins with a bifrontal craniotomy. Following that, the supraorbital rims and glabellar area with the lateral orbital walls are removed en bloc. In the temporal fossa, a tongue-and-groove arrangement provides stability. Both of these segments may require some modification to make the forehead less flat. They are then fixed together as described in the previous section and are then advanced as required.
Fixation is usually performed with metal miniplates in the temporal region because the resorbable plates tend to bend when the scalp is returned into its normal position. If closure becomes difficult because of the advancement, the whole scalp is mobilized posteriorly. If closure remains difficult, the galea is incised with multiple cuts from side to side. This yields satisfactory scalp expansion. [14] Because of their young age, these patients do not require bone grafts. Bone grafts are required in older patients, and a split-skull graft is the graft of choice. See the image below.
 Same patient following advancement, release of stenosis, and rearrangement of skull.
Same patient following advancement, release of stenosis, and rearrangement of skull.
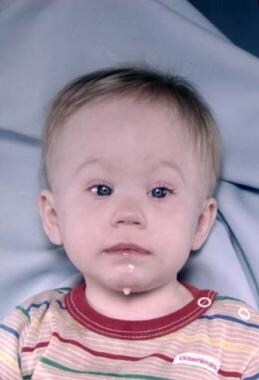
Trigonocephaly - Metopic Craniosynostosis
The midline forehead suture fusion results in a narrow anterior cranial fossa with a midline keel. Patients have hypotelorism with narrowing of the area between the medial canthi, and their orbits are rotated posterolaterally. [63] See the image below.
Treatment
A central coronal osteotomy splits the anterior cranial fossa and the supraorbital rims. The latter are rotated anteriorly and hinged medially, as are the frontal bones. This enlarges the anterior cranial fossa. The head is widened anteriorly. [64] See the images below.
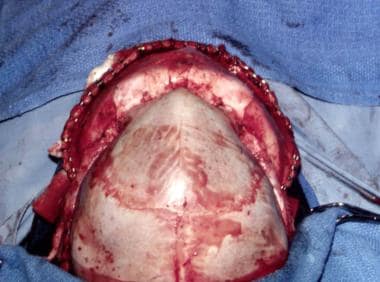 Metopic craniosynostosis. Appearance of skull from above. Note the central "keel" and the narrowing of the anterior cranial fossa.
Metopic craniosynostosis. Appearance of skull from above. Note the central "keel" and the narrowing of the anterior cranial fossa.
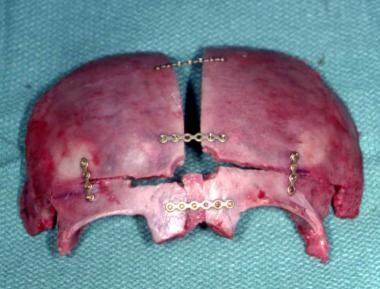 Metopic craniosynostosis. Expansion of the anterior cranial fossa and supraorbital area with insertion of bone grafts into the latter.
Metopic craniosynostosis. Expansion of the anterior cranial fossa and supraorbital area with insertion of bone grafts into the latter.
Acrocephaly or Turricephaly
This can result from multiple-suture fusion; thus, the skull grows upward. Basal suture fusion reduces the dimensions of the skull base.
Kleeblattschädel Anomaly
The characteristic of this deformity is the cloverleaf skull. [65] The vertex bulges upward, and the bitemporal regions bulge laterally. This gives the skull the appearance of having 3 lobes. These are superior anteriorly and bilateral in the temporal area. Variable degrees of this condition are reported. In some areas, the suture is closed; in others, the sutures may be wide open. The flow of cerebrospinal fluid seems to be interrupted in the region of the third ventricle. These children have a high prevalence of intellectual disability. This is also the case in children with Apert syndrome.
Treatment
This condition is treated similar to other craniosynostosis syndromes. The skull is exposed using a coronal flap, with a posterior coronal flap to widely expose the frontotemporal and occipital areas. All involved sutures are removed, and the individual areas of the skull are mobilized to adequately enlarge the skull. The deformed portions of the skull (eg, the temporal areas) are frequently recontoured by barrel-staving and molding. The bony segments are minimally stabilized to allow brain growth to proceed as normally as possible. Further releases may be required in the long term.
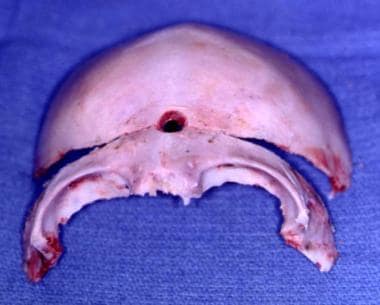
Sagittal Craniosynostosis - Scaphocephaly
This condition produces a long, narrow skull with some rotation of the supraorbital area such that the medial area is in advance of the lateral orbital region. [66, 67] See the images below.

Treatment
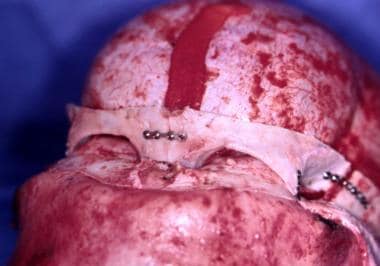
Two methods are available to address this situation. [68] A central strip of cranium can be removed from the coronal sutures to the occipital suture. See the image below.
However, if the surgeon is worried about the underlying sagittal sinus, strips of cranium can be removed from either side of the fused suture. Once this is performed, cuts are made anterior to the occipital suture and posterior to the coronal suture. The lateral skull segments are then out-fractured. If the frontosupraorbital area is unduly deformed as described earlier, it is removed en bloc and osteotomized in the midline to establish a proper frontosupraorbital contour. [61] These 2 procedures combined, when indicated, achieve adequate correction of this deformity. See the images below.
 Correction of sagittal craniosynostosis. Supraorbital area expanded with interpositional bone graft.
Correction of sagittal craniosynostosis. Supraorbital area expanded with interpositional bone graft.
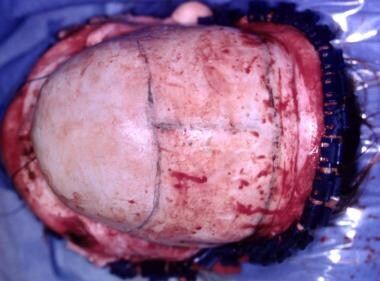
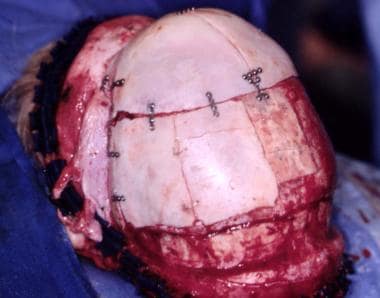 Correction of sagittal craniosynostosis. Skull is shortened anteroposteriorly and widened transversely.
Correction of sagittal craniosynostosis. Skull is shortened anteroposteriorly and widened transversely.
The occiput does not require correction because it is hidden by the hair, especially in females. If significantly involved, the posterior fossa may be enlarged by central and lateral osteotomies. This should be performed with care because the lateral sinuses can be transgressed, with disastrous results. See the images below.
Complications of Craniosynostosis Repair
In a study of 17,788 hospitalizations for surgical craniosynostosis repair, Allareddy found that complications arose in 10.1% of these, with hemorrhage (4.1%) being the most frequently reported complication; inhospital mortality was reported for 15 patients (0.08%). Most of the hospitalizations (77.7%) were for patients under age 1 year. Along with hemorrhage, the most frequently reported complications were as follows [69] :
-
Iatrogenically induced complications (3.1%): Including accidental punctures, lacerations, and pneumothorax
-
Bacterial infections (0.7%)
-
Cardiac complications (0.7%)
-
Respiratory complications (0.7%)
A literature review by Goyal et al reported that compared with open surgery for craniosynostosis repair, the endoscopic approach involves significantly less blood loss, operative time, and length of stay and significantly lower perioperative complication, reoperation, and transfusion rates. [70]
A study by Goldstein et al comparing complications of midfacial distraction osteogenesis using halo-type versus semiburied devices found a higher rate of operative repositioning in patients with the halo-type distractor, as a result of malposition or transcranial pin migration. However, patients with semiburied distractors experienced a higher rate of major infections. [71]
A study by Zimmerman et al found that although midface distraction osteogenesis in patients with craniosynostosis leads to acute worsening of velopharyngeal insufficiency (VPI), as measured using the Pittsburgh Weighted Speech Score (0.52 preoperatively versus 2.42 postoperatively), VPI improves, and often resolves, over time. [72]
-
Typical appearance of a patient with Crouzon syndrome, with maxillary retrusion, exorbitism, and pseudoprognathism. Anteroposterior view.
-
Typical appearance of a patient with Crouzon syndrome, with maxillary retrusion, exorbitism, and pseudoprognathism. Lateral view.
-
Crouzon syndrome. Anteriorly, the widened frontal and sagittal sutures can be seen.
-
Crouzon syndrome. The lateral view shows the erosion of the cranium by the gyri of the expanding brain.
-
The classic features of Apert syndrome, including the broad skull, bulging in the temporal area, and retrusion and vertical shortening of the maxilla.
-
The classic features of Apert syndrome, including the broad skull, bulging in the temporal area, and retrusion and vertical shortening of the maxilla.
-
Typical Apert syndrome hand with syndactyly of all fingers. The thumb has been freed surgically.
-
Child with Apert syndrome prior to correction.
-
Child with Apert syndrome prior to correction.
-
Surgical correction of Apert syndrome. This shows the complete correction of the frontosupraorbital and maxillary areas. Three separate segments are being moved independently and for different distances. Bony defects are filled with bone grafts.
-
Surgical correction of Apert syndrome. The frontoorbital area has been removed and has been repositioned and remolded. It is ready to be inserted.
-
Surgical correction of Apert syndrome. The bony segment is back in its advanced position, and the scalp is ready to be closed.
-
Same child 5 days after frontosupraorbital and maxillary advancement.
-
Same child 5 days after frontosupraorbital and maxillary advancement.
-
Child with Crouzon syndrome prior to correction.
-
Child with Crouzon syndrome prior to correction. Side view.
-
Appearance following frontosupraorbital advancement with simultaneous maxillary advancement at the Le Fort III level.
-
Appearance following frontosupraorbital advancement with simultaneous maxillary advancement at the Le Fort III level.
-
Adult with Crouzon syndrome.
-
Adult with Crouzon syndrome.
-
Postoperative appearance following Le Fort III and rhinoplasty.
-
Postoperative appearance following Le Fort III and rhinoplasty.
-
Correction of deformity from Crouzon syndrome using the rigid external distraction device. Preoperative appearance.
-
Correction of deformity from Crouzon syndrome using the rigid external distraction device. Preoperative appearance.
-
Osteotomies have been completed in the frontal and supraorbital areas. This complex has been advanced and fixed. Osteotomies have been performed on the maxilla, but it will be brought forward by external skeletal distraction.
-
Rigid external distraction device in position at the end of the procedure.
-
Postoperative distraction position. The dental occlusion is in an overcorrected position to allow for posterior relapse.
-
Postoperative distraction position. The dental occlusion is in an overcorrected position to allow for posterior relapse.
-
Pierre Robin syndrome. Preoperative appearance.
-
Pierre Robin syndrome. Preoperative appearance.
-
Pierre Robin syndrome. Distraction of mandible with intraoral device. The adjusting rod is seen perforating the skin.
-
Pierre Robin syndrome. Distraction of mandible with intraoral device. The adjusting rod is seen perforating the skin.
-
Pierre Robin syndrome. Postoperative result following mandibular distraction.
-
Pierre Robin syndrome. Postoperative result following mandibular distraction.
-
Treatment of hemicraniofacial microsomia. Preoperative appearance.
-
Treatment of hemicraniofacial microsomia. Preoperative appearance.
-
Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
-
Treatment of hemicraniofacial microsomia. Postoperative appearance. Zygomatic arch, mandibular reconstruction, and facial soft tissue augmentation with free bone graft, free vascularized iliac crest, and overlying soft tissue and skin.
-
Distraction device to lengthen mandible and ascending ramus.
-
Preoperative appearance showing atrophy of the right side of the face involving both soft tissue and bone.
-
Rectus abdominis free flap outlined and harvested.
-
Harvest of rectus abdominis free flap.
-
End result following reconstruction of right side of face with rectus myocutaneous free flap and bone grafting.
-
Treacher Collins syndrome. Preoperative appearance.
-
Treacher Collins syndrome. Preoperative appearance.
-
Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
-
Treacher Collins syndrome. Harvest of full-thickness skull to reconstruct lateral orbital rim and zygomatic arch.
-
Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
-
Treacher Collins syndrome. Zygomatic arch malar prominence and lateral orbital rim reconstructed with osteotomized full-thickness skull graft.
-
Same patient with Treacher Collins syndrome. Postoperative appearance.
-
Same patient with Treacher Collins syndrome. Postoperative appearance.
-
Preoperative appearance of patient with Treacher Collins syndrome.
-
Preoperative appearance of patient with Treacher Collins syndrome.
-
Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
-
Same patient. Postoperative result treated by the method outlined above. Distortion of the ears is due to bulky hearing aids.
-
Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
-
Hypertelorism. Preoperative appearance of patient with hypertelorism and maxillary retrusion.
-
Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
-
Hypertelorism. Diagram of osteotomies to be performed in the facial bipartition procedure. Involves resection of central bony block and rotation of the 2 halves of the face into a better position.
-
Hypertelorism. The ostectomy and osteotomies have been completed.
-
Hypertelorism. The ostectomy and osteotomies have been completed.
-
Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
-
Hypertelorism. The segments have been rotated into position. A cranial bone graft will be placed as shown in the diagram.
-
Hypertelorism. Postoperative result.
-
Hypertelorism. Postoperative result.
-
Patient with unilateral coronal craniosynostosis.
-
Unilateral coronal craniosynostosis. The frontal bone and the right supraorbital and lateral orbital wall have been removed.
-
Unilateral coronal craniosynostosis. The skull is rotated so that the flat part lies posteriorly on the opposite side.
-
Unilateral coronal craniosynostosis. The supraorbital rim is osteotomized.
-
Unilateral coronal craniosynostosis. Bone graft is inserted to make the right orbit the same size as the left orbit.
-
Appearance of the correction from above to show the bone graft in place.
-
Frequently, the supraorbital rim has to be rotated anteriorly as can be seen from this view.
-
Same patient. Postoperative result.
-
Patient with bicoronal craniosynostosis.
-
Same patient following advancement, release of stenosis, and rearrangement of skull.
-
Metopic craniosynostosis. Preoperative appearance.
-
Metopic craniosynostosis. Appearance of skull from above. Note the central "keel" and the narrowing of the anterior cranial fossa.
-
Metopic craniosynostosis. Expansion of the anterior cranial fossa and supraorbital area with insertion of bone grafts into the latter.
-
Same patient. Metopic craniosynostosis. Postoperative result.
-
Correction of sagittal craniosynostosis. Preoperative appearance.
-
Correction of sagittal craniosynostosis. Preoperative appearance.
-
Correction of sagittal craniosynostosis. Frontosupraorbital area removed.
-
Correction of sagittal craniosynostosis. Supraorbital area expanded with interpositional bone graft.
-
Correction of sagittal craniosynostosis. Plan of expansion of the skull.
-
Correction of sagittal craniosynostosis. Skull is shortened anteroposteriorly and widened transversely.
-
Same patient. Correction of sagittal craniosynostosis. Postoperative appearance.
-
Same patient. Correction of sagittal craniosynostosis. Postoperative appearance.
Tables

What would you like to print?
- Crouzon, Apert, Pfeiffer, Saethre-Chotzen, and Carpenter Syndromes
- Pierre Robin Sequence
- Hemifacial Deformity
- Romberg Disease (Progressive Hemifacial Atrophy)
- Klippel-Feil and Turner Syndromes
- Treacher Collins Syndrome (Mandibulofacial Craniosynostosis)
- Hypertelorism and Hypotelorism
- Craniosynostosis
- Show All
- Media Gallery
- References




