Practice Essentials
Cornelia de Lange syndrome (CdLS) is a syndrome of multiple congenital anomalies characterized by a distinctive facial appearance, prenatal and postnatal growth deficiency, feeding difficulties, psychomotor delay, behavioral problems, and associated malformations that mainly involve the upper extremities. Molecular genetic diagnosis can be performed using sequencing and deletion/duplication analysis of the genes NIPBL, SMC3, RAD21, SMC1A, and HDAC8. Early intervention in patients with Cornelia de Lange syndrome is necessary for feeding problems, hearing and visual impairment, congenital heart disease, and urinary system abnormalities.
Cornelia de Lange first described it as a distinct syndrome in 1933, [1] although Brachmann had described a child with similar features in 1916. [2] Diagnosing classic cases of Cornelia de Lange syndrome is usually straightforward; however, diagnosing mild cases may be challenging, even for an experienced clinician. See the images below.
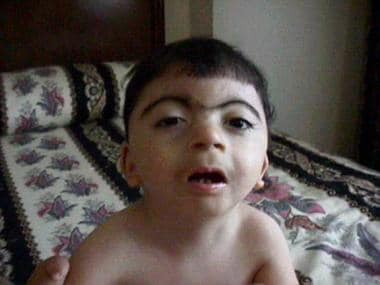 Facial appearance of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
Facial appearance of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
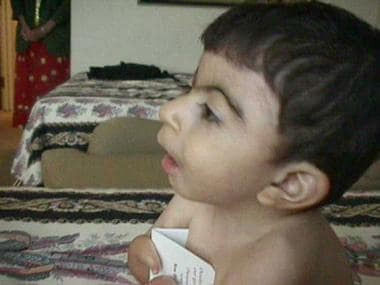 Facial profile of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
Facial profile of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
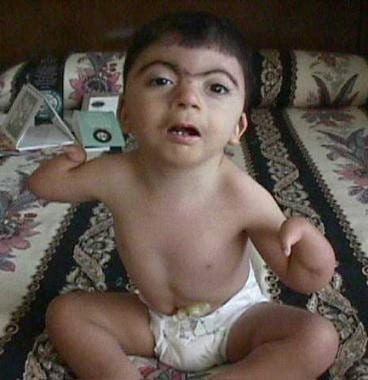 Severe upper-extremity malformations in a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
Severe upper-extremity malformations in a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
Signs and symptoms of Cornelia de Lange syndrome
These include the following:
-
Intrauterine growth retardation (68%)
-
Prematurity (31%)
-
Low-pitched, weak cry in infancy - Noted in classic cases and disappears as the child grows (74%)
-
Initial hypertonicity (100%)
-
Respiratory and feeding difficulties in the newborn period and infancy (71%)
-
Developmental delay and intellectual disability
-
Seizures (23%) with no specific electroencephalographic (EEG) pattern
-
Behavioral manifestations - Including hyperactivity (40%), self-injury (44%), daily aggression (49%), and sleep disturbance (55%)
Workup in Cornelia de Lange syndrome
Molecular genetic diagnosis can be performed using sequencing and deletion/duplication analysis of NIPBL, SMC3, RAD21, SMC1A, and HDAC8.
This testing can confirm the diagnosis of Cornelia de Lange syndrome, especially in mild or atypical cases, and the results can help in identifying the family specific mutation for prenatal testing in future pregnancies.
Radiography can reveal bone abnormalities, along with hiatal hernia, aspiration pneumonia, gastroesophageal reflux, and intestinal obstruction, malrotation, and volvulus.
Ultrasonography can be used to assess for kidney and urinary tract abnormalities, voiding cystourethrography is indicated for evaluation of recurrent urinary tract infections or hydronephrosis, and echocardiography is indicated for evaluation of congenital heart disease.
Radiologic brain findings may include enlarged ventricles, including enlargement of basal cisterns; thinning or atrophy of white matter, particularly frontal lobes, with relative sparing of parietal lobes; brainstem hypoplasia; and cerebellar vermian hypoplasia or agenesis.
Management
Early intervention in patients with Cornelia de Lange syndrome is necessary for feeding problems, hearing and visual impairment, congenital heart disease, and urinary system abnormalities.
Early intervention for psychomotor delay is also indicated. Computer programs that emphasize visual memory are more beneficial than standard methods of verbal instruction. Perceptual organizational tasks should be emphasized. Tactile stimulation during indirection helps the children remember and perform maximally.
Surgery may be necessary for the following conditions:
-
Cleft palate
-
Nasal polyps
-
Gastroesophageal reflux disease
-
Pyloric stenosis
-
Intestinal malrotation/volvulus
-
Undescended testis
-
Lacrimal duct stenosis
-
Hip dislocations
Pathophysiology
More than 99% of cases are sporadic. Cornelia de Lange syndrome is occasionally transmitted in an autosomal dominant pattern, according to several instances in which a usually mildly affected parent had one or more affected offspring. Twins with concordance and discordance have been reported. Although possible autosomal recessive inheritance has been reported in some families, these instances were likely to be due to germline mosaicism. [3] The recurrence risk is 0.5-1.5% if parents are unaffected and 50% if a parent is affected.
Heterozygous mutations in a gene named NIPBL, the human homolog of the Drosophila melanogaster Nipped-B gene, [4] have been identified in approximately 50% of individuals with Cornelia de Lange syndrome. [5, 6, 7] Although the exact function of the protein product of NIPBL in humans (delangin) remains unknown, its homologs in other species are known to play roles in developmental regulation and in cohesion of sister chromatids. Mutations in genes, coding for two other proteins involved in cohesion of sister chromatids, SMC1A and SMC3, have been reported in 5% and 1% of patients with Cornelia de Lange syndrome, respectively. [8] Thus, Cornelia de Lange syndrome is considered to be a cohesinopathy, along with Roberts syndrome/SC phocomelia.
Inheritance is autosomal dominant in families with NIPBL and SMC3 mutations and is X-linked dominant in families with SMC1A mutations.
All types of NIPBL mutations, including missense, splice-site, nonsense, and frameshift mutations, have been reported to result in the Cornelia de Lange syndrome phenotype. The most likely effect of these mutations is haploinsufficiency. The mutation-detection rate is approximately 50%. Genomic deletions and duplications of the NIPBL locus are rare. [9, 10] Reported mutations of SMC1A include missense mutations and in-frame deletions. One reported SMC3 mutation is an in-frame deletion.
The correlation between genotype and phenotype suggested that individuals with an identifiable mutation in NIPBL have a phenotype more severe than the phenotype of those without mutations. Moreover, missense mutations in NIPBL are associated with mild phenotypic features. Patients with mutations in SMC1A and SMC3 consistently have a milder phenotype, with absence of severe limb defects and other structural anomalies. The phenotype in some patients is close to those with nonsyndromic intellectual disability.
A study by Gil-Rodríguez et al of SMC3 -associated phenotypes in 16 patients found a tendency toward the following [11] :
-
Less distinctive syndrome-related craniofacial features (although, as in other cases of Cornelia de Lange syndrome, microcephaly is characteristic)
-
Fewer associated congenital heart defects
-
Normal limbs (as mentioned above)
-
Milder prenatal growth retardation (although it becomes more severe in childhood)
A phenotype similar to that of Cornelia de Lange syndrome may be observed in patients with a duplication of band q26-27 on chromosome 3. [12] Molecular studies of genes mapped to this region of chromosome arm 3q have failed to reveal mutations in patients with Cornelia de Lange syndrome.
Some autopsy data have indicated cerebral dysgenesis, with a decreased number of neurons, neuronal heterotopias, and focal gyral folding abnormalities, as causes of psychomotor delay.
Kaur et al showed that expression levels of NIPBL, as well as other genes, correlate with the phenotypic severity in Cornelia de Lange syndrome. Nonsense mutations in NIPBL led to about 35% reduced expression and gave rise to severe phenotypes. Missense and inframe deletions in NIPBL and other genes were associated with 20% reduced expression and produced milder phenotypes. [13]
Epidemiology
Frequency
The incidence is 1 case per 10,000-50,000 live births. No differences based on sex or race have been described.
Mortality/Morbidity
Gastrointestinal disease complications are one of the most common causes of death in this syndrome. They include diaphragmatic hernia in infancy and aspiration pneumonia and volvulus at an older age.
A retrospective review of 295 propositi with Cornelia de Lange syndrome found that respiratory issues (eg, aspiration, reflux, pneumonia) were the most common causes of death (31%), followed by gastrointestinal disease, including obstruction/volvulus (19%), congenital anomalies (15%), neurological causes, (8%), accidents (8%), sepsis (4%), acquired cardiac disease (3%), cancer (2%), renal disease (1.7%), and other causes (9%). [14]
Special Concerns
A prenatal diagnosis is made after Cornelia de Lange syndrome (CdLS)-related abnormalities are carefully evaluated using prenatal ultrasonography. These abnormalities include growth retardation, limb defects, diaphragmatic hernia, hypoplastic forearms, underdeveloped hands, and typical facial defects.
The availability of molecular diagnosis should substantially improve prenatal diagnosis. Prenatal diagnosis with molecular genetic techniques is currently available if a mutation is known in the family.
Failure to detect a mildly affected parent may result in incorrect risk estimation for future pregnancies.
Anatomic abnormalities of the face and neck may cause difficulties during intubation. GI obstruction or feeding difficulties may occur. Early feeding management is important.
Severe speech delay and poor communication are concerns. The patient may have congenital heart disease.
Prognosis
Very few cases of adults with Cornelia de Lange syndrome have been reported in the literature. Two cohorts of adults, totaling 122 patients, have been studied. These individuals ranged in age from 15-50 years, with an average age of 17 and 24. Premature aging was seen among these patients. The most common medical issues included the following [15] :
-
Gastroesophageal reflux diseases (GERD) - 71-82%
-
Overweight/obesity - 16-31%
-
Limb length discrepancy - 26-46%
-
Epilepsy - 26%
-
Hearing loss - 42-65%
-
Vision problems - 38-50%
-
Behavioral issues - 50%
All individuals reported having not required continuing assistance with nasogastric tubes and were able to feed orally in adulthood.
Epilepsy in Cornelia de Lange syndrome has proven to be amenable to medications, while cardiac issues typically do not require surgery in adulthood (18%). Intellectual disabilities are variable, as follows:
-
Borderline - 11%
-
Mild - 29%
-
Moderate - 18%
-
Severe - 16%
-
Profound - 19%
A prognostic index created by Cereda et al for Cornelia de Lange syndrome uses a point system to help predict the severity of a child's intellectual disabilities early in development. [16]
Patient Education
Information can be obtained from the Cornelia de Lange Syndrome Foundation, Inc., at http://www.cdlsusa.org/.
-
Facial appearance of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
-
Facial profile of a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
-
Severe upper-extremity malformations in a patient with Cornelia de Lange syndrome. Courtesy of Ian Krantz, MD, Children's Hospital of Philadelphia.
Tables
Parameter |
1 point |
2 point |
3 point |
Birth weight |
Above 2,500 g |
2,000–2,500 g |
Below 2,000 g |
Sitting alone |
< 9 mo |
9–20 mo |
>20 mo |
Walking alone |
< 18 mo |
18–42 months |
>42 mo |
Saying first word |
< 24 mo |
24–48 mo |
>48 mo |
Upper limb malformation |
No defect |
Partial defect (>2 digits) |
Severe defect (< 2 digits) |
Number of other major malformations |
0-1 |
2-3 |
>3 |
Hearing loss |
Absent |
... |
... |
A score of less than 15 points indicates mild involvement, a score of 15-22 points indicates moderate involvement, and a score of more than 22 points indicates severe involvement. |
|||


