Practice Essentials
Acid sphingomyelinase deficiency (ASMD) is a rare lipid storage disorder with a genetic etiology. It is commonly known as Niemann-Pick disease (NPD) type A, type B, or type A/B. The original description of NPD referred to what is currently termed NPD type A, which is a fatal disorder of early childhood characterized by failure to thrive, hepatosplenomegaly, and a rapidly progressive neurodegenerative course that leads to death by age 2-3 years.
Since this original description, other forms of NPD have been described. NPD type B (chronic visceral ASMD) is a milder, non-neuronopathic form with later onset and longer survival, sometimes into adulthood. NPD types A and B are inherited as autosomal recessive traits.
NPD type A/B is an overlapping disease entity that is also known as chronic neurovisceral ASMD or NPD B variant. It has later symptom onset compared with type A and slower neurologic and visceral disease progression compared with type A and type B.
NPD type C, a rarer form, results from defects in cholesterol metabolism, specifically defects in two distinct cholesterol-binding proteins (NPC1 and NPC2), [1] rather than from ASMD. Hence, type C will not be discussed here.
Signs and symptoms of sphingomyelinase deficiency
Niemann-Pick disease (NPD) type A
The first sign detected is usually the presence of hepatosplenomegaly, which is evident at age 3 months and becomes progressively greater.
Psychomotor development does not progress beyond the 12-month level, and in later stages, the affected child is completely unable to interact with the environment.
Mild hypotonia may be evident by age 6 months, followed by progressive loss of tone and deep tendon reflexes. With disease progression, loss of motor function occurs, and in the final stages, spasticity and rigidity are evident.
Affected infants exhibit feeding problems, failure to thrive, recurrent respiratory tract infections, and irritability. Most children with NPD type A die before age 2-3 years, often from respiratory failure following pulmonary infection.
Niemann-Pick disease (NPD) type B
The condition is diagnosed in most patients in infancy or childhood when enlargement of the liver, spleen, or both is detected during routine physical examination. At diagnosis, patients with NPD type B also have evidence of mild pulmonary involvement.
Recurrent pneumonia and life-threatening bronchopneumonias may occur, and cor pulmonale has been described. Severely affected patients may also have liver involvement leading to life-threatening cirrhosis, portal hypertension, and ascites.
Workup in sphingomyelinase deficiency
The diagnosis of NPD is confirmed with measurement of enzyme activity in peripheral white blood cells or in cultured fibroblasts.
Targeted mutation analysis for four common SMPD1 mutations is available in clinical laboratories.
The pathologic hallmark in NPD types A and B is the histochemically characteristic lipid-laden foam cell, often termed the Niemann-Pick cell, on bone marrow examination.
Pancytopenia may be present secondary to the enlarged spleen in patients with NPD, and reduced high-density–lipoprotein cholesterol (HDL-C) fraction is common in NPD type B.
Chest radiography in NPD type B reveals a typical reticulonodular pattern of infiltration, even in patients with no overt pulmonary symptoms. Calcified pulmonary nodules may be seen. Skeletal imaging in NPD type B often shows delayed bone age.
Computed tomography (CT) scanning or magnetic resonance imaging (MRI) in NPD type B can be used to quantify liver and spleen volumes. MRI of the brain in NPD type A may be normal or may show signs of white matter involvement.
Management of sphingomyelinase deficiency
The first disease-specific enzyme replacement treatment for non–central nervous system (non-CNS) ASMD, olipudase alfa, was approved by the US Food and Drug Administration (FDA) in August 2022.
Other medical and surgical care for NPD types A and B are mainly focused on supportive care.
Adult patients with elevated serum cholesterol due to NPD type B should be treated to bring serum cholesterol concentration into the normal range. Liver function should be monitored in patients treated with statins.
Transfusion of blood products may be necessary for acute episodes of bleeding secondary to hypersplenism and thrombocytopenia in NPD type B patients.
Supplemental oxygen may be used for NPD type B patients with symptomatic interstitial lung disease. Bronchopulmonary lavage has had variable results.
Pathophysiology
Niemann-Pick disease (NPD) types A and B result from genetic mutations in the SMPD1 gene, producing in a deficiency of acid sphingomyelinase (ASM) and lysosomal accumulation of sphingomyelin.
The SMPD1 gene spans about 5 kb of chromosome 11 (11p15.1-11p15.4) and consists of six exons. Its 1896 bp frame encodes a 631 amino-acid protein in the endoplasmic reticulum. This pre-pro polypeptide undergoes cleavage and preprocessing in the endoplasmic reticulum and Golgi complex, resulting in the 70 kDa mature form of ASM localized to lysosomes; this is further processed to a 52 kDa form. [2]
ASM enzyme activity is necessary for breaking down sphingomyelin into ceramide and phosphocholine. Most patients with NPD type A typically have less than 5% residual ASM enzyme activity, while those with NPD type B may have over 10% enzyme activity. [3] Certain mutations, such as p.F572L, may result in a residual enzyme activity of about 30%, but due to dramatic reduction of the protein half-life, the condition may phenotypically be type A. [2] Since residual enzyme activity and phenotype are not always correlated, residual enzyme activity should not be used as a predictor of phenotype; rather, it should serve as an adjunct to molecular and clinical assessment in the diagnosis and treatment of NPD. [4]
The enzymatic defect results in the pathologic accumulation of sphingomyelin (which is a ceramide phospholipid) and other lipids in the monocyte-macrophage system, the primary site of pathology in patients with NPD.
In addition to sphingomyelin, bis(monoacylglycero)phosphate and lyso-sphingomyelin are also elevated. Cholesterol, glucocerebroside, lactosylceramide, and gangliosides (particularly GM3) are elevated as well, but not to the extent seen in NPD type C.
Sphingomyelin is a major component of cell membranes and the principal phospholipid of the myelin sheath. [1, 5] (See the image below.)
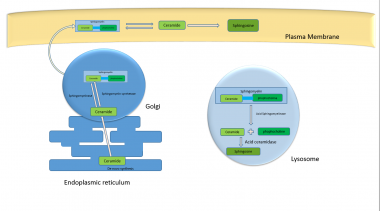
Progressive deposition of sphingomyelin in the central nervous system (CNS) results in the neurodegenerative course observed in NPD type A, which shares systemic disease manifestations with NPD type B. The severity of systemic involvement—which includes progressive lung disease, hepatosplenomegaly, short stature, and pancytopenia—varies in affected individuals.
Etiology
Niemann-Pick disease (NPD) types A and B result from deficient activity of sphingomyelinase, a lysosomal enzyme encoded by the SMPD1 gene, located on bands 11p15.1-p15.4. [6, 7] To date, more than 180 mutations have been described. [2]
Various types of mutations have been reported, including, most frequently, missense mutations (65.3%), with others including frameshift mutations (19%), nonsense mutations (7%), frame deletions, intronic variants, mutant alleles, duplication, and indel mutations. Large deletions or insertions have not been reported. [2]
Frameshift mutations usually result in little or no enzyme activity and typically produce the type A phenotype. With missense mutations, significant residual activity is retained, so such mutations are related to the type B phenotype. [3, 8] Although, as previously mentioned, the missense mutation pF572L results in 30% enzyme activity, it also dramatically reduces protein half-life and is therefore associated with the type A phenotype. [2]
The complete sphingomyelinase genomic region has been isolated and sequenced. Three common mutation variants (p.R498L, p.L304P, p.P333Sfs*52) are associated with NPD type A. The delta-R608 mutation is a common mutation that results in NPD type B. In addition, p.P323A, p.P330R, and p.W393G variants are associated with NPD type B. Mutation variants p.Q294K and p.W393G are associated with an intermediate phenotype. [4]
Regional distribution of mutations causing NPD varies. In the Maghreb region of North Africa, delta R608 may account for almost 90% of mutant alleles, resulting in NPD type B, while in the Turkish population, three mutations—L137P, fsP189, and L549P—are responsible for over 70% of cases of type B. [9] In addition, a study reported that the mutations c.409T>C (p.L137P) and c.567delT (p.P189fsX65) were found in 40% and 35% of alleles, respectively, in the entire Turkish population. [3] In the Saudi Arabian population, the H421Y and K576N mutations account for 85% of NPD type B cases. Increased prevalence of distinct mutations has also been described in various other ethnic groups, including Scottish/British and Brazilian/Portuguese populations. [9]
SMPD1 gene defects have also been reported as strong risk factors for Parkinson disease, with variants p.L304P and p.R591C having particularly been associated with such risk in the Ashkenazi Jewish and Chinese populations. [2]
Epidemiology
Race
Niemann-Pick disease (NPD) is a rare disorder that occurs in persons of all races. [10] Frequency and distribution of mutations are variable in different ethnic groups and even between different families in the same ethnic groups.
NPD type A is more common in persons of Ashkenazi Jewish descent. Guidelines for carrier screening for genetic disorders in this population have been established. [11]
NPD type B occurs worldwide but is more common in the Maghreb region of North Africa, in Saudi Arabia, and in individuals of Turkish descent. NPD type B has various phenotypes, and they are found commonly in Arab, Turkish, and Portuguese populations. In North African patients with NPD type B who originated from Maghreb region, the delta-R608 mutation accounts for almost 90% of mutant alleles. [9]
Sex
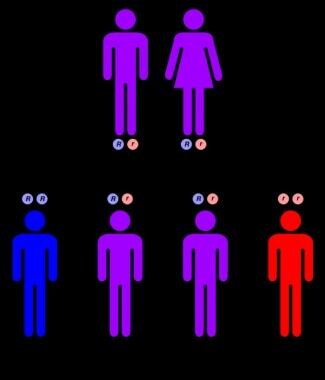
NPD types A and B are autosomal recessive disorders (see image below), equally affecting males and females.
Age
Patients with NPD type A disease develop symptoms as early as age 3 months. NPD type B has a variable age of presentation but frequently appears early in childhood, when hepatosplenomegaly is detected and symptoms of lung involvement may occur.
Prognosis
Mortality/morbidity
In Niemann-Pick disease (NPD) type A, the clinical presentation and course are relatively uniform and characterized by normal appearance at birth and hepatosplenomegaly from age 3 months. Development does not progress beyond age 12 months; a relentless neurodegenerative course then follows.
Patients with NPD type A usually die by age 2-3 years. In contrast, patients with type B disease often survive to adulthood, although most present in childhood with hepatosplenomegaly.
Patients with NPD type B primarily have visceral involvement, sometimes massive. In contrast to the stereotyped type A phenotype, the clinical presentation and course of patients with NPD type B disease are more variable with respect to clinical findings, age of onset, and severity of symptoms. Most patients are diagnosed in infancy or childhood, when enlargement of the liver and the spleen are detected during routine physical examination.
A study by Cassiman et al found that in patients with chronic neurovisceral ASMD (intermediate NPD A/B; NPD B variant), the most frequent causes of death were neurodegenerative disease (23.1%), respiratory disease (23.1%), and liver disease (19.2%). Death in patients with chronic visceral ASMD (ASMD type B), however, mainly resulted from respiratory disease (30.9%), liver disease (29.1%), and bleeding (12.7%). [12]
Complications
Complications of Niemann-Pick disease (NPD) include the following:
-
Patients with NPD type B are at risk for splenic rupture and should avoid contact sports
-
A small number of patients with NPD type B develop liver failure and may be candidates for liver transplantation
-
Pulmonary disease is progressive in patients with NPD type B and may result in oxygen dependence
-
Coronary artery or valvular heart disease can occur in NPD types B and A/B
Patient Education
Counsel families regarding genetic risk and the availability of prenatal testing.
For patient education resources, see the Cholesterol Center, as well as High Cholesterol, Cholesterol FAQs, and Cholesterol Drugs
-
Autosomal recessive inheritance pattern.
-
Role of acid sphingomyelinase in metabolism pathways inside cellular organelles.






