Practice Essentials
Fabry disease is an X-linked lysosomal storage disease that is caused by deficient activity of lysosomal enzyme α-galactosidase A (α-Gal A). Most males with no α-Gal A activity develop the classic phenotype of Fabry disease, which affects multiple organ systems. The first clinical manifestations of the disease, which consist of episodes of severe pain in the extremities (acroparesthesias), hypohidrosis, corneal and lenticular changes, and skin lesions (angiokeratoma), develop in childhood. [1] See the images below.
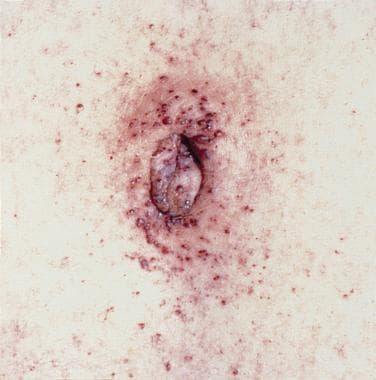 Angiokeratoma is the small punctate reddish-to-bluish angiectases on the umbilicus.
Angiokeratoma is the small punctate reddish-to-bluish angiectases on the umbilicus.
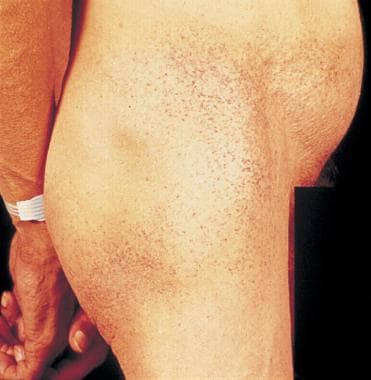 Angiokeratomas are commonly observed as dense cluster of lesions on the flank and private areas.
Angiokeratomas are commonly observed as dense cluster of lesions on the flank and private areas.
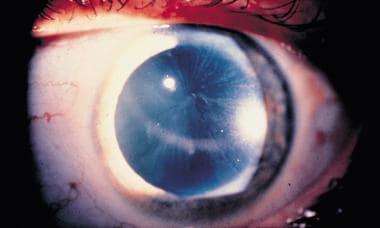 Corneal verticillata, commonly seen in patients with Fabry disease, detectable by slit lamp examination.
Corneal verticillata, commonly seen in patients with Fabry disease, detectable by slit lamp examination.
The rate of disease progression and specific organ damage demonstrate intrafamilial and interfamilial variability. Renal failure, cardiovascular disease, and stroke are the major causes of morbidity and mortality, occurring in the fourth or fifth decade of life.
Signs and symptoms of Fabry disease
Early manifestations include the following:
-
Corneal and lenticular opacities
-
Skin lesions (angiokeratomas)
-
Pain in the extremities (acroparesthesia)
-
Decreased ability to sweat (hypohidrosis)
In addition, patients may have chronic abdominal pain and diarrhea.
During the late stage of the disease, the progressive deterioration of renal, cardiac, and nervous system function ultimately results in significant morbidity and mortality.
Late and serious manifestations include the following:
-
Paroxysmal attacks of severe rotational vertigo
-
Sensorineural hearing loss
-
Poor heat and exercise tolerance
-
Transient ischemic attacks
-
Vascular thromboses
-
Seizures
-
Hemorrhagic or ischemic stroke
-
Angina pectoris
-
Variant angina
-
Myocardial infarction
Workup in Fabry disease
Laboratory studies in Fabry disease include the following:
-
α-Gal A
-
DNA analysis
-
Complete blood count (CBC)
-
Serum electrolyte level measurement
-
Lipid profile
-
Renal evaluation - Serum blood urea nitrogen (BUN) and creatinine levels and 24-hour urine or spot urine measurement for total protein/creatinine, albumin/creatinine, sodium, creatinine, and urinary GL-3 (optional) levels [2]
Imaging studies include the following:
-
Magnetic resonance imaging (MRI) - To document evidence of brain ischemic disease
-
Magnetic resonance angiography (MRA) - May be indicated to assess cerebral vasculopathy
Other tests include the following:
-
Echocardiography - To demonstrate ventricular hypertrophy and septal thickening
-
Electrocardiography (ECG) - Findings can include sinus bradycardia, nonspecific ST-segment changes, T-wave inversion, and shortened PR interval
-
Pulmonary evaluation - Induced sputum analysis, lung biopsy, or both if severe pulmonary involvement is present (to exclude an intercurrent disease process)
-
Slit-lamp microscopy - To identify the typical Fabry disease–specific changes in the cornea, lens, retina, and conjunctiva
-
Psychosocial evaluation
Management of Fabry disease
Fabry disease management strategies should be tailored to the individual according to patient age and disease stage. These strategies include the use of medication to alleviate the symptoms, disease-specific treatment to delay and prevent possible serious organ damage, and adherence to standard health-care measures and a healthy lifestyle.
With regard to pain management, daily prophylactic doses of neuropathic pain agents (eg, phenytoin, carbamazepine, gabapentin, or a combination of these agents) provide some degree of relief. Some patients may require more potent analgesics (eg, opioids) for pain management.
A growing body of evidence suggests that enzyme replacement therapy (ERT) is beneficial in improving most disease symptoms. However, the response to ERT may vary, depending in part on tissue-specific differences in drug delivery and disease stage.
Pathophysiology
Glycosphingolipids, predominantly globotriaosylceramide (GL-3) and galabiosylceramide, accumulate in the lysosomes of various cells (eg, in the vascular endothelium of multiple organs) owing to α-Gal A deficiency. The accumulation of GL-3 in the lysosomes causes lysosomal and cellular dysfunction; this, in turn, triggers the cascade of cells and tissue ischemia and fibrosis.
Epidemiology
Frequency
United States
Fabry disease is one of the more common lysosomal storage disorders, affecting approximately 1 in 40,000-60,000 males.
Mortality/Morbidity
Prior to the availability of renal transplant, dialysis, and, more recently, enzyme replacement therapy (ERT), the average age at death in men with classic Fabry disease was 41 years. Renal failure, heart failure and/or myocardial infarction, and stroke were among the most likely causes of death.
Race
Although most patients with Fabry disease are white, the disorder has been described in patients in many ethnic groups, including those with Hispanic, African, Asian, and Middle Eastern ancestry.
Sex
As is expected in X-linked disorders, males with deleterious mutations have little to no residual α-Gal A activity. Therefore, these patients experience the full spectrum of disease symptoms. Because of random X inactivation (lyonization), the disease presentation in female carriers is more variable and depends on the normal-to-mutant ratio of α-Gal A in the different tissues. A significant number of female carriers may develop Fabry disease–related symptoms, including acroparesthesias, GI symptoms, renal and cardiac disease, and/or stroke.
Age
Most males with classic Fabry disease first manifest symptoms in childhood or early adolescence. The earliest manifestations include acroparesthesias, angiokeratomas, hypohidrosis, and lenticular and corneal changes. Proteinuria usually becomes evident in the second decade of life, and renal insufficiency is typically present in the third decade of life. Cardiovascular and cerebrovascular diseases usually develop in the fourth decade of life.
Individuals with atypical renal or cardiac variants usually do not have signs or symptoms in childhood. Many of these patients remain asymptomatic well into adulthood, when patients with classic symptoms are severely affected or have died from the disease.
-
Angiokeratoma is the small punctate reddish-to-bluish angiectases on the umbilicus.
-
Angiokeratomas are commonly observed as dense cluster of lesions on the flank and private areas.
-
Corneal verticillata, commonly seen in patients with Fabry disease, detectable by slit lamp examination.






