Background
Double aortic arch is one of the most common forms of vascular ring, a class of congenital anomalies of the aortic arch system in which the trachea and esophagus are completely encircled by connected segments of the aortic arch and its branches. Although the double aortic arch has various forms, the common defining feature is that both the left and right aortic arches are present.
Embryology
The easiest way to understand the anatomy and development of double aortic arch and other forms of vascular ring is to begin by considering the bilateral system of pharyngeal arch vessels that exists in early embryogenesis.
Early in the course of embryonic morphogenesis, six pairs of pharyngeal arch arteries develop in conjunction with the branchial pouches. The first six arches appear in a fairly sequential fashion, with left-to-right symmetry and constitute the primitive vascular supply to the brachiocephalic structures, running from the aortic sac to the paired dorsal aortas. As normal cardiovascular morphogenesis proceeds, a patterned regression and persistence of the various arches and right-sided dorsal aorta occur, ultimately resulting in the mature configuration of the thoracic aorta and its branches. The third, fourth, and sixth arches, along with the seventh intersegmental arteries and the left dorsal aorta, are the primary contributors to the normal aortic arch and its major thoracic branches (see diagram below).
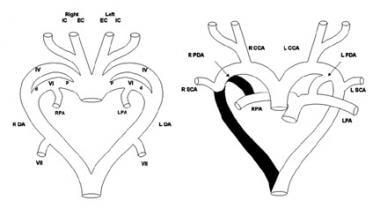 Double Aortic Arch. Schematic diagram (left) of the primitive pharyngeal arch system shows the left (L) and right (R) external carotid (EC) and internal carotid (IC) arteries, fourth (IV) and sixth (VI) pharyngeal arches, distal pulmonary arterial segments (PA), dorsal aortas (DA), and seventh intersegmental arteries (VII). The proximal (p) sixth arches develop into the proximal pulmonary arteries and the distal (d) sixth arches become the arterial ducts. The seventh intersegmental arteries develop into the subclavian arteries. Schematic diagram (right) shows the segments of the pharyngeal arch system that regress (shown in black) in the normal formation of the thoracic great arteries. Left pulmonary artery (LPA); ductus arteriosus (PDA); right pulmonary artery (RPA); subclavian artery (SCA).
Double Aortic Arch. Schematic diagram (left) of the primitive pharyngeal arch system shows the left (L) and right (R) external carotid (EC) and internal carotid (IC) arteries, fourth (IV) and sixth (VI) pharyngeal arches, distal pulmonary arterial segments (PA), dorsal aortas (DA), and seventh intersegmental arteries (VII). The proximal (p) sixth arches develop into the proximal pulmonary arteries and the distal (d) sixth arches become the arterial ducts. The seventh intersegmental arteries develop into the subclavian arteries. Schematic diagram (right) shows the segments of the pharyngeal arch system that regress (shown in black) in the normal formation of the thoracic great arteries. Left pulmonary artery (LPA); ductus arteriosus (PDA); right pulmonary artery (RPA); subclavian artery (SCA).
The segments of the bilateral aortic arch system that normally regress include the distal portion of the sixth arch and the right-sided dorsal aorta. Normally, the left fourth arch becomes the aortic arch, the right fourth arch contributes to the innominate artery, the distal left sixth arch becomes the ductus arteriosus, the proximal sixth arches bilaterally contribute to the proximal branch pulmonary arteries, the left dorsal aorta becomes the descending thoracic aorta, and the dorsal intersegmental arteries bilaterally become the subclavian arteries.
Vascular rings are formed when this process of regression and persistence does not occur normally, and the resulting vascular anatomy completely encircles the trachea and esophagus. [1] (Other forms of aortic arch anomalies occur in which a vascular ring is not present.) A double aortic arch is formed when both fourth arches and both dorsal aortas remain present. The reasons for the absence of regression are not clearly known.
Anatomy
Double aortic arch has various forms. Both arches may be patent, or an atretic (but persistent) segment may be present at one of several locations in either arch. When both arches are patent, the right or left arch may be larger, or they may be similar in size. A cervical arch on either side, variable laterality of the descending thoracic aorta, coarctation of the major arch, and/or discontinuity of the central pulmonary arteries may be present. In general, the apex of the right-sided arch is more superior than the left arch, and on occasion, a cervical arch may be present on either side.
In more than 75% of patients with double aortic arch, the right arch is dominant. Among patients with a right-dominant double arch, those with a patent minor arch outnumber those with an atretic minor arch. When the minor arch is atretic, the atretic segment almost always is distal to the left subclavian artery, although atresia may also occur between the left common carotid and subclavian arteries. In approximately 20% of patients, the left arch is dominant. In these patients, the minor right arch is typically patent.
Associated cardiovascular anomalies
Double aortic arch usually occurs without associated cardiovascular anomalies. Ventricular septal defect and tetralogy of Fallot are probably the most common associated defects, although truncus arteriosus, transposition of the great arteries, pulmonary atresia, and complex univentricular defects sometimes occur in conjunction with a double arch.
Associated syndromes and noncardiac conditions
Double aortic arch is associated with a chromosome 22q11 deletion in approximately 20% of patients (see Etiology). Chromosome 22q11 deletion is responsible for DiGeorge, velocardiofacial, and conotruncal anomaly face syndromes, which are often referred to using the unified terms CATCH-22 syndrome or chromosome 22q11 deletion syndrome. In patients with double aortic arch, the frequency of phenotypes satisfying the clinical criteria for these various syndromes is not known. Rather, the important point is that double aortic arch may be associated with chromosome 22q11 deletion, which has various other possible manifestations. These include, but are not limited to, palatal abnormalities, laryngotracheal anomalies, speech and learning delay, characteristic facial features, hypocalcemia, abnormalities of T-cell–mediated immune function, and neurologic defects.
Occasionally, patients with double aortic arch may have anomalies consistent with either vertebral, anal, cardiac, tracheal, esophageal, renal, and limb (VACTERL) or posterior coloboma, heart defect, choanal atresia, retardation, genital, and ear (CHARGE) associations. Double aortic arch has also been reported in association with other chromosomal anomalies, such as trisomy 21 and other syndromes.
One of the more important noncardiac features that sometimes is found in association with double aortic arch is esophageal atresia, insofar as an undiagnosed arch anomaly may complicate repair of the esophageal atresia, which is usually recognized earlier than the double aortic arch.
Another noncardiac anomaly that may be associated with vascular rings is a congenital laryngeal web, which may present with the same symptoms and signs as a vascular ring. Accordingly, patients with persistent stridor or upper airway obstruction after repair of a vascular ring, particularly those with a chromosome 22q11 deletion, should be evaluated for the presence of a congenital laryngeal web.
Pathophysiology
By definition, a vascular ring encircles the trachea and esophagus, usually causing compression of both structures. Compression of the trachea causes upper airway obstruction that impairs inspiratory and, to a lesser degree, expiratory airflow. [2] The extent of respiratory impairment depends on the severity of compression, which can widely vary. Significant compression of the trachea appears to be more common with double aortic arch than with other forms of vascular ring and is often more severe.
In addition to airway symptoms, patients may experience swallowing difficulties related to esophageal compression. These typically manifest as vomiting and feeding intolerance in infants and younger children and as dysphagia later in life. Swallowing dysfunction may contribute to respiratory symptoms as a result of aspiration and/or compression or irritation of the membranous portion of the trachea as a food bolus traverses the area of esophageal obstruction. Although respiratory or esophageal pathophysiology may predominate in any given patient, respiratory compromise usually is more problematic in younger patients. Patients with primarily esophageal symptomatology tend to be older at presentation. [3] The pathophysiology of double aortic arch does not differ in the various anatomic forms.
Etiology
A persistent double aortic arch occurs when abnormal regression of the embryonic aortic arch segments is present, in which both the left and right aortic arches remain intact. With the different forms of double aortic arch, different segments of the embryonic aortic arch system, which normally regress, remain patent.
Factors responsible for the aberrant persistence of certain aortic arch segments have not been clearly identified, and the pathogenesis of this anomaly remains a mystery. Double aortic arch typically occurs without associated cardiovascular defects, although other lesions may be present, and accordingly, it is not usually found as part of a syndromic complex.
In one study, chromosome 22q11 deletions were found in 3 of 22 patients (14%) with double aortic arch. [4] This chromosomal anomaly is associated with aortic arch anomalies in patients with other forms of conotruncal heart disease and other isolated vascular abnormalities, and chromosome 22q11 deletion is likely to be an important etiologic factor in double aortic arch. Most such mutations arise de novo, and no recognizable inheritance pattern is present.
Familial recurrence of double aortic arch has been reported, supporting a genetic etiology for this anomaly. Teratogen-induced double aortic arch in animal models also has been reported. The mechanisms and significance of these models have not been elucidated.
Epidemiology
United States data
Generally, incidence of double aortic arch and vascular rings is unknown, although vascular rings comprise an estimated 1% of cardiovascular malformations that are managed surgically. In most surgical series, 45-65% of patients undergoing repair of a vascular ring have a double aortic arch.
Race-, sex-, and age-related demographics
Based on limited data, no racial predilection is apparent.
No sex predilection has been documented in patients with double aortic arch or its various subtypes.
Double aortic arch is a developmental abnormality that is present in the fetus. The postnatal age at which the anomaly is identified may vary, although in most patients, double aortic arch is diagnosed in early infancy. Double aortic arch may also be identified on prenatal ultrasonography. [5]
Prognosis
Long-term prognosis for patients with repaired double aortic arch is excellent; persistent respiratory symptoms are the most common adverse outcomes.
In patients with a repaired double aortic arch, lifestyle implications are minimal and most likely are related to residual symptoms or associated anomalies.
Morbidity/mortality
The natural history of double aortic arch is not well defined. Vascular rings were among the first congenital cardiovascular anomalies repaired surgically, and surgical management has been the standard of care for more than 50 years. Patients with significant airway compression may die as a result of respiratory compromise, but such events are rare.
Preoperative morbidity generally is limited to respiratory symptoms, feeding problems, or both. Some patients may develop recurrent respiratory infections, and some may exhibit failure to thrive as the result of a combination of increased metabolic requirements from respiratory and feeding work and relatively poor oral intake.
Complications
Complications are uncommon after repair of vascular rings.
The major postoperative symptom is persistent respiratory symptoms, including cough, dyspnea, and wheezing. Pulmonary function testing reveals persistent upper airway obstruction in some patients. Others have evidence of lower airway obstruction that usually is responsive to bronchodilator therapy. Whether the incidence of lower airway obstruction is higher in patients who have undergone repair of vascular rings than in the population at large or whether such a pathologic condition in patients with rings has any relationship to prior anatomic and functional abnormalities is not known.
Rarely, patients with unrepaired double aortic arch may develop an aortoesophageal fistula, which causes fatal hemorrhage. Another rare complication is esophageal erosion.
-
Double Aortic Arch. Schematic diagram (left) of the primitive pharyngeal arch system shows the left (L) and right (R) external carotid (EC) and internal carotid (IC) arteries, fourth (IV) and sixth (VI) pharyngeal arches, distal pulmonary arterial segments (PA), dorsal aortas (DA), and seventh intersegmental arteries (VII). The proximal (p) sixth arches develop into the proximal pulmonary arteries and the distal (d) sixth arches become the arterial ducts. The seventh intersegmental arteries develop into the subclavian arteries. Schematic diagram (right) shows the segments of the pharyngeal arch system that regress (shown in black) in the normal formation of the thoracic great arteries. Left pulmonary artery (LPA); ductus arteriosus (PDA); right pulmonary artery (RPA); subclavian artery (SCA).
-
Double Aortic Arch. Schematic diagram (left) depicts the segments of the pharyngeal arch system that regress (shown in black) so that the mature vascular anatomy of a double aortic arch can develop. The dominant and minor arches can vary in laterality and specific patterns of branching and segmental hypoplasia/atresia. (These variables are not specified in this diagram.) Left (L) and right (R) external carotid (EC) and internal carotid (IC) arteries; fourth (IV) and sixth (VI) pharyngeal arches; distal pulmonary arterial segments (PA); dorsal aortas (DA); seventh intersegmental arteries (VII); proximal (p) sixth arches; distal (d) sixth arches. Mature anatomy (right) of a double aortic arch with a dominant right arch and patent minor left arch. In most patients, a single left-sided ductus arteriosus or ligamentum arteriosum is present. Left pulmonary artery (LPA); ductus arteriosus (PDA); right pulmonary artery (RPA); subclavian artery (SCA).
-
Double Aortic Arch. Transverse MRI images in a patient with double aortic arch. Both arches are patent; the right arch is dominant. Images A-F are arranged in a caudad to cephalad order. (A) Transverse image at the level of the pulmonary valve. The ascending aorta (AAo) and descending aorta (DAo), cephalad to the junction of the left and right arches, can be seen. (B) At the level of the pulmonary artery (PA) bifurcation, the distal confluence of the left and right arches forming the single descending aorta is depicted. (C) The distal portions of the left (L) and right (R) arches can be seen posterior and to the left and right sides of the trachea. Note the anteroposterior compression of the tracheal carina (anterior to and between the arches). (D) Moving cephalad, the dominance of the right arch can be seen. (E) At the level of the proximal/transverse aortic arches, the origin of the left and right arches from the rightward ascending aorta can be seen. (F) The left and right common carotid and subclavian arteries arise from the left and right arches, respectively. The common carotid arteries are the dark round structures anterior to and to either side of the trachea. The subclavian arteries are the dark round structures posterior to and to either side of the trachea.
-
Double Aortic Arch. Coronal spin-echo MRI images in a patient with a double aortic arch. Both arches are patent, with the right (R) slightly larger in caliber than the left (L). Compression of the trachea (T) between the two arches can be seen (left). The confluence of the arches and the descending aorta (D) are shown (right).





