Overview
von Hippel-Lindau (VHL) disease, or von Hippel-Lindau syndrome, is a rare genetic disorder characterized by visceral cysts and benign tumors in multiple organ systems that have subsequent potential for malignant change. [1]
Clinical hallmarks of VHL disease include the development of retinal and central nervous system (CNS) hemangioblastomas (blood vessel tumors), pheochromocytomas, multiple cysts in the pancreas and kidneys, and an increased risk for malignant transformation of renal cysts into renal cell carcinoma. The wide age range and pleiotropic manner in which VHL disease presents complicates diagnosis and treatment in affected individuals, as well as their at-risk relatives. The image below illustrates a hemangioblastoma of the retina as found in patients with VHL disease. [1]
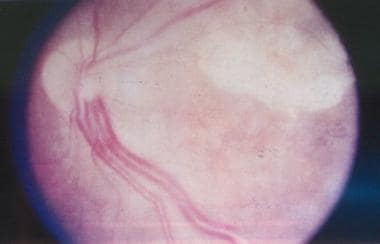 von Hippel-Lindau disease. Clinical picture of the retina, showing a pair of dilated and tortuous feeder vessels coursing on the surface of the retina from the optic nerve head toward the angioma (which is not seen in this image because it is in the extreme periphery).
von Hippel-Lindau disease. Clinical picture of the retina, showing a pair of dilated and tortuous feeder vessels coursing on the surface of the retina from the optic nerve head toward the angioma (which is not seen in this image because it is in the extreme periphery).
Hemangiomas
VHL disease is characterized by retinal capillary hemangiomas (also called benign vascular hamartomas). [2] Diagnosed in 50% of patients with VHL disease, these hemangiomas are composed of endothelial cells and pericytes. The foamy stromal cells between the capillaries stain positive for glial fibrillary acid protein and neuron-specific enolase. [3]
Retinal capillary hemangiomas, usually supplied by large dilated feeder vessels, may occur in any part of the retina. Serum leakage from these vessels and hemangiomas leads to retinal exudates. Organized fibroglial bands with traction retinal detachment and vitreous hemorrhage may occur, along with potential complications such as glaucoma or permanent vision loss. [4]
Tumors involving other organs and the CNS (brain, spinal cord) are present in 25% of patients with VHL disease. [5] Pancreatic and renal cysts can also occur and may be present concurrently.
Diagnostic considerations
The unexpected finding of a retinal or CNS hemangioblastoma or the diagnosis of a pheochromocytoma should prompt a search for other associated VHL disease features, as many of these patients may have the diagnostic criteria for VHL disease. Early identification of VHL is important because of the increased risk of serious complications (eg, renal cell carcinoma) to foster more effective treatment options and better prognoses.
Because VHL disease is a multiple-organ disease that widely varies in clinical presentation, various manifestations may lead to diagnosis. Criteria are the following:
-
More than one hemangioblastoma in the CNS (brain, spinal cord) or eye
-
A single hemangioblastoma in the CNS or retina, plus a visceral manifestation (multiple renal, pancreatic, or hepatic cysts; pheochromocytoma; renal cancer)
-
Positive family history plus any one of the above clinical manifestations
-
Elucidation of a deleterious mutation in the VHL gene
Genetic testing for mutations in the VHL gene is performed at many laboratories throughout the United States and the world. Gene Tests (www.genetests.org) cites 48 different laboratories in the United States that can test for the VHL gene mutation. Some of these locations are as follows:
-
Boston University School of Medicine, Center for Human Genetics, Boston, Massachusetts
-
Children's Mercy Hospital, Molecular Genetics Laboratory, Kansas City, Missouri
-
Johns Hopkins Hospital, DNA Diagnostic Laboratory, Baltimore, Maryland
-
University of Pennsylvania School of Medicine, Genetic Diagnostic Laboratory, Philadelphia, Pennsylvania
Epidemiology
VHL disease is inherited in an autosomal-dominant Mendelian pattern. De novo mutations occur in about 1:4.4 million live births and account for 20% of cases. The incidence of VHL disease in the United States is approximately 1 case in 36,000 live births (worldwide incidence is 1:32,000 live births). Males and females are equally affected, and the diagnosis is made in people of all ethnic groups.
Age at diagnosis varies from infancy to age 60-70 years, with an average patient age at clinical diagnosis of 26 years. Clinically significant issues typically arise in affected individuals who are in their teens or twenties. About 20% of children with VHL can have ocular or adrenal signs before age 10 years. People also can first present with clinical findings in their ninth decade of life.
Physiology
The von Hippel-Lindau (VHL) gene (VHL) is located on the short (p) arm of chromosome 3 (3p25.3) and encodes a ubiquitously expressed 4.7 kilobase (kb) messenger ribonucleic acid (mRNA) that encodes 3 alternately spliced exons. The resultant 2 encoded von Hippel-Lindau protein (pVHL) products, a 30-kD full-length form (p30) and a 19-kD form (p19), shuttle between the nucleus and the cytoplasm, where they form complexes with other proteins. These protein complexes have many functions: the regulation of senescence, the oxygen-sensing pathway, microtubule stability and orientation, cilia formation, cytokine signaling, collagen IV assembly into the extracellular matrix, assembly regulation of a normal extracellular fibronectin matrix, and tumor suppression. [6]
To some extent, the ubiquitous expression of pVHL explains the pleiotropic manifestations of von Hippel-Lindau (VHL) disease. At present, mutational changes in the VHL gene are the only known cause of VHL disease. Molecular genetic testing of VHL gene detects mutations in 90%-100% of affected individuals. [7]
Functions of von Hippel-Lindau protein
Polyubiquitination
pVHLs form a complex with several other proteins (elongins B and C, Cullin2, Rbx1). This multiprotein complex ubiquitinates different substrates, thus marking them for degradation. pVHL gives this complex its target specificity, recruiting specific proteins to the complex for degradation. Two key targets recruited to the protein complex under normal oxygen conditions are the transcription factors HIF1a (hypoxia-inducible factor-1a) and HIF2a (hypoxia-inducible factor-2a). Ubiquitinated substrates are normally degraded, which means that defective pVHL leads to the accumulation of undegraded products in the body.
Regulation of transcription factors: HIF1a and HIF2a
Cells lacking pVHL fail to degrade HIFs in the presence of oxygen, thus permitting accumulation of high levels of stable proteins and activating the transcription of a large cohort of hypoxia-responsive genes constitutively. Although many tumors have high levels of HIF genes in hypoxic regions of the tumor, tumors resulting from pVHL inactivation express high levels of HIF genes in all of the tumor cells.
Regulation of other hypoxia-inducible genes
Cells without pVHL activity overproduce other hypoxia-induced mRNAs, such as those for erythropoietin (EPO), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), and enzymes involved in glycolysis through HIF-mediated transcription. Tumors associated with VHL disease are often highly vascular, possibly because of overproduction of these mRNAs. Paraneoplastic polycythemia is frequently observed as a result of increased EPO production.
Interaction with the extracellular matrix
pVHL also binds to microtubules and to fibronectin, a glycoprotein that interacts with structural proteins of the cell. Cells with defective pVHL have increased proliferation and decreased differentiation.
Cell cycle control
Control of the cell cycle is likely a multifactorial activity; pVHL can interact with cyclin D1 and affects the exit from the cell cycle.
Tumorigenesis
Understanding how the loss of pVHL function causes tumorigenesis is not fully known. The VHL gene may act as a classic tumor suppressor gene, as originally described by Knudson in his 2-hit theory of carcinogenesis. [8] When this theory is applied to an individual with VHL disease, the person inherits a germline mutation that renders one VHL allele inactive and an acquired "second hit" to the other VHL allele in a somatic cell leaves that cell without tumor suppressor ability. Application of Knudson's theory leads to a selective growth advantage and an increased risk of malignant progression.
Prognosis
Owing largely to the high incidence of clear cell renal cell carcinoma associated with von Hippel-Lindau (VHL) disease, the average life expectancy in affected individuals is 49 years. However, diligent surveillance enabling early treatment interventions can increase life expectancy. The morbidity of VHL disease varies depending on the particular organ system involved and the extent of the organ-system insult.
Belzutifan (Welireg), a hypoxia-inducible factor inhibitor, was approved by the FDA in August 2021. It is indicated for treatment of adults with VHL disease who require therapy for associated RCC, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery. Belzutifan binds to hypoxia-inducible factor 2 alpha (HIF-2α) and prevents HIF-2α from interacting with hypoxia-inducible factor 1 beta, resulting in reduced transcription and expression of HIF-2α target genes.
Renal lesions
The primary cause of morbidity and mortality in VHL disease, as well as the most serious sequela of the condition, involves the malignant degeneration of renal cysts. Renal cysts seldom are clinically significant; however, in VHL disease, they have a substantial rate of malignant transformation. Approximately 40% of patients with VHL disease develop this complication. Other renal lesions, such as hemangiomas and benign adenomas, can occur.
Renal cell carcinoma (namely, clear cell renal cell carcinoma) is the leading cause of death in patients with VHL disease, with a prevalence of as high as 75% reported in one autopsy series. The average age at which patients with VHL disease develop renal cell carcinoma is 44 years, about 20 years earlier in life than the age at which sporadic renal cell carcinoma is diagnosed in the general population. Renal cell carcinoma is the presenting finding in approximately 10% of patients diagnosed with VHL disease. The risk of developing renal cell carcinoma increases with age, so that by age 60 years, the risk is approximately 70% in individuals with VHL disease. Hence, these facts reinforce the importance of obtaining routine renal imaging studies.
Diagnostic radiological imaging of the kidneys (via ultrasonography, computed tomography [CT] scanning, or magnetic resonance imaging [MRI]) is mandatory in patients with VHL disease and at-risk relatives. Annual renal imaging using ultrasonography should be initiated in the teenage years. In older patients, CT scanning and/or MRI is used. Radiologists with expertise in these various imaging modalities are critical for proper interpretation.
A nephron-sparing treatment approach, such as tumor excision and/or partial nephrectomy, is recommended to try to preserve renal function in patients with renal cell carcinoma. Surgical intervention is considered once the tumor size reaches 3 cm. [9] Patients with VHL disease have a high incidence of tumor regrowth; as such, bilateral nephrectomy may ultimately be required, necessitating dialysis or renal transplantation for survival.
Targeted therapy, specifically antiangiogenesis, is another option in patients with VHL disease who have clear cell renal carcinoma. The goal of antiangiogenesis therapy is to halt the process of creating new blood vessels. Necessary nutrients needed to sustain tumor growth are delivered through the vascular system. The intent of antiangiogenesis is to “starve” the tumor tissue by preventing blood flow delivery. An antiangiogenic drug called sunitinib, marketed as Sutent by Pfizer, is an oral medication that is a multi-targeted receptor tyrosine kinase (RTK) inhibitor. In January 2006, this drug was approved by the US Food and Drug Administration (FDA) for the treatment of advanced renal cell carcinoma in patients with VHL disease. [9, 10]
CNS hemangioblastomas
CNS hemangioblastomas are the second most common cause of morbidity and mortality in patients with VHL disease. Approximately 70% of patients with VHL disease develop these tumors, with the mean age of diagnosis being 25 years. These CNS blood vessel tumors typically arise below the tentorium. Eighty percent of these tumors originate in the cerebellum and 20% in the spine.
Radiographically, these hypervascular masses are best detected using gadolinium-enhanced MRI. Although hemangioblastomas are usually benign, enlargement of these tumors within the confines of the CNS can cause neurologic compromise. Surgical resection may be a treatment option in this situation.
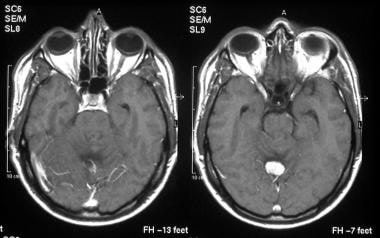 T1-weighted, transaxial, gadolinium-enhanced magnetic resonance imaging (MRI) scans show a well-defined, hypervascular, enhancing mass.
T1-weighted, transaxial, gadolinium-enhanced magnetic resonance imaging (MRI) scans show a well-defined, hypervascular, enhancing mass.
Retinal hemangioblastomas
Retinal hemangioblastomas (also called retinal angiomas) are diagnosed in patients with VHL disease at an average age of 29 years. Although patients with retinal hemangioblastomas are usually asymptomatic, these benign ocular tumors can lead to considerable morbidity through retinal detachment or vision loss from an enlarging lesion. In addition, patients with retinal angiomas have a 25% risk of developing CNS hemangioblastomas, primarily in the cerebellum. [11] Loss of vision is caused by lipid exudate in the macula area. Early detection of retinal angiomas with prompt medical attention can prevent significant vision loss. Successful treatments using diathermy, laser, or cryocoagulation have been used. Untreated retinal angiomas can lead to retinal detachment. Subsequent potential complications include vitreous hemorrhage, secondary iris neovascularization with glaucoma, and cataract formation.
Ocular complications associated with retinal angiomas are as follows:
-
Macular exudation
-
Retinal detachment
-
Vitreous hemorrhage
-
Cataract
-
Glaucoma
-
Nerve damage
-
Iatrogenic complications from argon laser photocoagulation, cryotherapy, or irradiation
Endolymphatic sac tumors
Endolymphatic sac tumors (ELSTs) of the middle ear are vascular lesions growing within the posterior temporal bone. [12] They often occur bilaterally and are diagnosed in about 10% of patients with VHL disease. Presenting clinical signs include hearing loss, tinnitus, vertigo, and facial weakness. Hearing loss of various degrees is the major complication if appropriate surgical intervention is not performed.
Pheochromocytomas
Patients with VHL disease are at an increased risk for developing pheochromocytomas. These neuroendocrine tumors of adrenal gland tissue are usually histologically benign. Clinical presentation may be asymptomatic or may cause episodic or sustained hypertension in patients with VHL disease. Often seen in younger patients (median age, 30 years), these tumors are often multiple or involve extra-adrenal tissue. Only about one third of patients have increased catecholamine production.
Screen patients and at-risk family members for the presence of pheochromocytomas with standard biochemical means. When detected, the treatment approach is the same as for patients with sporadic pheochromocytomas not associated with the VHL clinical spectrum.
A patient’s risk of developing pheochromocytomas hinges on the precise nature of the mutation responsible for VHL disease in their family. In kindreds with VHL disease that have a deletion or protein-truncating mutation of the VHL gene (type 1 VHL disease), the lifetime risk for developing pheochromocytoma is less than 10%. Patients with large deletions in the VHL gene that include the HSPC300 gene have a subtype of type 1 VHL disease. This subtype, called VHL type 1B, protects patients from developing renal cell carcinoma. [13]
In patients within kindreds having a missense mutation as their genetic etiology for VHL disease (type 2 VHL disease), the risk for developing pheochromocytoma increases to approximately 50%. Type 2 VHL disease can be subdivided into subtypes 2A, 2B, and 2C. Each subtype has a relative risk for the development of renal cell carcinoma in VHL patients presenting with pheochromocytoma. Patients with type 2A have a low risk of developing renal cell carcinoma, whereas those with type 2B have a high risk. Patients with type 2C have an increased risk for pheochromocytoma, but not for renal cell carcinoma.
Different VHL types and subtypes can be distinguished based on predominant pathology in reference to pheochromocytomas, as follows [14] :
-
VHL type 1 - Renal carcinoma and hemangioblastoma
-
VHL type 2A - Hemangioblastoma and pheochromocytoma
-
VHL type 2B - Renal carcinoma and pheochromocytoma
-
VHL type 2C - Pheochromocytoma only
Pancreatic lesions
Pancreatic involvement is common in patients with VHL disease. Most (70%) pancreatic lesions are simple cysts and rarely cause symptoms or develop into malignant tumors. Neuroendocrine tumors of the pancreas are less common and have malignant potential, with risk of metastasis to the liver. Malignant islet cell tumors, functioning islet cell tumors, or pancreatic carcinomas can occur in individuals with VHL disease. Abdominal imaging should be performed regularly to assess for pancreatic lesions.
Epididymal papillary cystadenomas
Epididymal papillary cystadenomas are present in about 50% of male patients with VHL disease. These benign cysts are usually asymptomatic and do not require treatment. Single papillary cystadenomas occur in the general population. This isolated finding in a patient does not pose concern for VHL disease if other salient disease findings are absent. However, bilateral epididymal cystadenomas are pathognomonic for VHL disease.
An equivalent benign lesion in females is a papillary cystadenoma of the ovarian broad ligament. Symptoms may include pain, dyspareunia, and menorrhagia. Symptomatic treatment is indicated in these women.
Physical Examination
Retinal hemangioblastomas (retinal angiomas)
Approximately 50% of patients with von Hippel-Lindau (VHL) disease have retinal hemangioblastomas. These lesions are visible with direct ophthalmoscopic evaluation. [15] They appear as a dilated artery leading from the disc to a peripheral tumor with an engorged vein. Patients usually present with retinal hemangioblastomas in their third decade of life.
von Hippel-Lindau disease. Clinical picture of the retina, showing a pair of dilated and tortuous feeder vessels coursing on the surface of the retina from the optic nerve head toward the angioma (which is not seen in this image because it is in the extreme periphery).
Endolymphatic sac tumors
As mentioned above, presenting clinical signs for ELSTs include hearing loss, tinnitus, vertigo, and facial weakness. If the patient is not treated within a timely fashion to receive appropriate surgical intervention, hearing loss of varying severity can be a major complication.
Differential Diagnosis
Conditions to consider in the differential diagnoses of von Hippel-Lindau (VHL) disease include the following:
-
Multiple endocrine neoplasia, type 2 (MEN type 2)
-
Multiple paraganglioma syndrome
-
Neurofibromatosis
-
Pheochromocytoma
-
Autosomal dominant polycystic kidney disease (ADPKD)
-
Birt-Hogg-Dube (BHD) syndrome
-
Tuberous sclerosis complex (TSC)
-
Choroidal mass (tumor/metastasis)
-
Retinal telangiectasia
-
Retinal macroaneurysm
Retinal and CNS hemangioblastomas are pathognomonic for VHL disease, and their presence is diagnostic. These lesions also clearly differentiate VHL disease from similar diagnoses also being considered by the medical care team.
Multiple endocrine neoplasia, type 2
A diagnosis of pheochromocytoma should prompt diagnostic consideration of VHL disease and MEN type 2. MEN type 2 is easily differentiated from VHL disease based on the presence of parathyroid tumors and medullary thyroid carcinoma.
Multiple paragangliomas syndrome
Multiple paragangliomas are observed in patients with mutations in the succinate dehydrogenase (SDH) complex gene, subunits B (SDHB), C (SDHC), and D (SDHD). [16] Paragangliomas are endocrinologically active tumors of the sympathetic nervous system that are histologically identical to pheochromocytomas. These paragangliomas occur in the head and neck region, which distinguishes them from the typical adrenal-tissue locations of pheochromocytomas. Paragangliomas are notpart of clinical spectrum of VHL disease.
Autosomal-dominant polycystic kidney disease
Multiple renal cysts are found in individuals with ADPKD, and these cysts are much more plentiful than what is observed in the renal tissue of persons with VHL disease. In ADPKD, the renal architecture is markedly distorted, adversely affecting renal function. The risk for malignant change is low in persons with ADPKD.
CNS lesions are seen in both ADPKD and VHL disease; however, the lesions in ADPKD consist of arterial aneurysms, not hemangioblastomas, as seen in VHL disease. Hepatic cysts are common in ADPKD but rare in VHL disease, whereas pancreatic cysts are rare in ADPKD and common in VHL disease.
Birt-Hogg-Dube syndrome and renal cell carcinoma
In BHD syndrome, a predisposition for renal cell carcinoma is seen. BHD syndrome is inherited as a Mendelian autosomal dominant and is due to a mutation in the folliculin (FLCN) gene located on chromosome 17p11.2. Malignancies in these patients are not due to the known VHL gene located on chromosome 3p25.3. Regardless, patients diagnosed with renal cell carcinoma require proper medical investigation to search for the presence of other clinical abnormalities.
Tuberous sclerosis complex
Tuberous sclerosis complex (TSC) needs to be considered in the differential diagnoses of patients presenting with multiple renal lesions. Renal cysts occur in both TSC and VHL disease; however, renal tumors typically seen in TSC are angiomyolipomas, which have a characteristic appearance on abdominal CT scanning and MRI. Unlike VHL disease, TSC is a neurocutaneous disorder characterized by neurologic and dermatologic findings. Salient dermatologic findings of TSC are hypopigmented macules (ash-leaf spots), shagreen patches, periungual fibromas, and adenoma sebaceum. Seizure disorders and learning disabilities are also part of the TSC clinical spectrum.
Laboratory Studies
Manifestations of von Hippel-Lindau (VHL) disease are pleiotropic. In the early stages, most aspects of the disorder can be detected using radiographic imaging studies, biochemical analyses, or both.
Laboratory studies should be performed annually for the following patients:
-
Patients diagnosed with VHL disease
-
Patients in whom VHL disease is suspected
-
Kindred members (relatives) who are at risk for VHL disease
Recommended blood and urine tests are discussed below.
Complete blood cell (CBC) count is used to look for evidence of polycythemia vera due to EPO expression by renal cysts and cerebellar hemangioblastomas.
Electrolytes and renal function (BUN and creatine) are used for electrolyte measurement and renal baseline function.
Measurement of plasma catecholamines and urinary catecholamine metabolites (inclusive of vanillylmandelic acid [VMA], fractionated metanephrines [especially normetanephrines], and total catecholamines) is performed to detect pheochromocytomas, even when hypertension is absent. Screening for pheochromocytomas and paragangliomas should include measurements of plasma-free metanephrines or urinary fractionated metanephrines.
The 2014 Endocrine Society Guidelines for obtaining sera and urine samples are as follows:
-
For a blood sample, patients should be supine for 20-30 minutes from the time the needle is inserted and when blood is drawn. Catecholamine release by peripheral nerves and adrenal glands is stimulated by upright-seated posture, causing an increase in plasma metanephrines when compared to the supine position of blood sampling.
-
Blood sample analysis should use reference standards from supine tests, not seated tests.
-
Urine collection should be over a 24-hour period. [17]
-
Urinalysis is performed for hematuria, which can indicate a renal abnormality.
-
Urine cytology is used to detect findings that suggest renal cell carcinoma.
Imaging Studies
Imaging studies used to diagnose von Hippel-Lindau (VHL) disease include the following:
-
Ophthalmic ultrasonography
-
Abdominal/genitourinary ultrasonography: Renal and pancreatic lesions, cysts of the epididymis and broad ligament
-
Abdominal CT scanning without and with contrast: Renal, pancreatic, and adrenal gland lesions
-
Abdominal MRI: Renal, pancreatic, and adrenal gland lesions
-
Brain CT scan with and without contrast
-
CNS MRI with and without contrast
Medical personnel must be cautious of contrast use in patients with VHL disease who have renal impairment. [7]
Ophthalmic imaging
Ocular color Doppler ultrasonography is a safe and noninvasive investigative modality used in diagnostic ophthalmology. This technique provides a simultaneous morphologic and vascular image of the disease entity, enabling diagnosis and monitoring the effectiveness of treatment. A 7.5-MHz linear array transducer (Acuson 128XP) is used to examine the eyes. Real-time grayscale images and color Doppler images are obtained. Pulsed Doppler analysis also can be used to evaluate vascular dynamics.
In clinically diagnosed cases of retinal angiomatosis, color Doppler ultrasonography is used to delineate the dilated, enlarged, and closely running feeder vessels on the retinal surface. The image below illustrates Doppler findings highly suggestive of retinal angiomatosis. [18, 19]
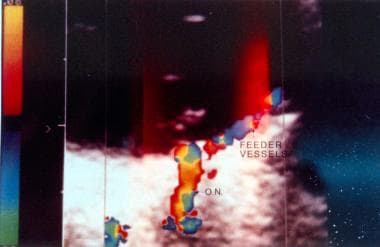 von Hippel-Lindau disease. Color Doppler image of the dilated feeder vessels with flow patterns in opposite directions as indicated by red for the artery and blue for the vein. The abbreviation ON designates the optic nerve with its blood vessels.
von Hippel-Lindau disease. Color Doppler image of the dilated feeder vessels with flow patterns in opposite directions as indicated by red for the artery and blue for the vein. The abbreviation ON designates the optic nerve with its blood vessels.
This color Doppler ultrasonography shows the feeder vessels to be a pair of dilated tortuous blood vessels coursing to the angioma from the optic disc. The color image illustrates flow in these vessels to be in opposite directions; however, no flow can be appreciated in the angioma. These Doppler findings are of great value in diagnosing retinal angiomatosis especially when visualization of the fundus is not possible with an ophthalmoscope owing to opaque media, a potential complication of the angiomatosis.
Regression of the ocular disease can be monitored with repeated ocular ultrasonographic studies after various treatment modalities (ie, cryotherapy, diathermy, irradiation, laser photocoagulation). It is common to see new angiomas appearing or growing in the region of treated angiomas.
Optical coherence tomography (OCT) and neuroimaging advances have made the ocular changes present in VHL disease more visible, allowing for earlier interventions and better outcomes. [20]
Abdominal imaging
Annual abdominal imaging studies are recommended in the following persons:
-
Patients diagnosed with VHL disease
-
Individuals in whom VHL disease is suspected
-
At-risk relatives of a patient diagnosed with VHL disease
Abdominal ultrasonography can be used to identify lesions in the kidneys, adrenal glands, and/or pancreas. CT scanning of the abdomen, with and without contrast, is recommended for additional clarification of abdominal lesions. However, MRI may be of equal or greater value, depending on the expertise of the medical personnel performing and interpreting the imaging studies.
Renal imaging
The American College of Radiology has established guidelines for the assessment of indeterminate renal masses. [21] These guidelines address which imaging techniques are ideal to further clarify renal masses, a critical resource for patients with VHL disease.
CNS imaging
Periodic imaging to detect hemangioblastomas of the brain and spinal cord is usually not required. These CNS tumors are typically benign; therefore, imaging is reserved for patients presenting with neurologic symptoms. Focal neurologic signs should immediately prompt imaging of the brain and spinal cord. MRI is the preferred imaging modality owing to the preponderance of subtentorial and posterior fossa tumors.
CNS MRI imaging should include the brain and cervical, thoracic, and lumbar spine. MRI scans should be performed as a 1.5T MRI with and without contrast, with thin cuts through the posterior fossa. Attention should be given to the inner ear/petrous temporal bone to rule out ELSTs and hemangioblastomas of the neuraxis.
Evaluation of At-Risk Family Members
von Hippel-Lindau (VHL) disease is inherited as a Mendelian autosomal-dominant trait; hence, the children of affected patients have a 50% risk of inheriting the disorder. However, the degree of clinical severity varies and cannot be predicted in each affected family member. Siblings, parents, and extended family members all have potential risk of having VHL disease. As with other autosomal-dominant disorders, there is a risk for new or de novo mutations (ie, no mutation is identified in either parent) to occur for the first time in an individual. For VHL disease, this de novo mutation occurs in about a 20% of patients.
The genetic origin of the disease’s heritability enables a search for the causative mutation in the proband or the initially identified patient. This ability greatly benefits at-risk family members. If the proband's causative mutation can be identified, its presence or absence in at-risk family members can reliably and unequivocally define their status for development of VHL disease. Individuals who have not inherited the mutated VHL allele can manage without the burden and recommended task of annual screening, and individuals with the mutated allele can be closely monitored for early signs of VHL disease.
Genetic testing for mutations in the VHL gene requires complete sequencing of the coding regions and is approximately 80% sensitive. The addition of Southern blot analysis detects virtually all mutations. In the rare event that no causative mutation is identified in the proband, all at-risk relatives must continue to undergo annual medical assessment and appropriate screening until age 60 years.
Interpretation of pedigree information and gene mutational analysis is complicated. The assistance of personnel trained in medical genetics (geneticists, genetic counselors) is needed to give correct guidance to patients and their family members.
Nonsurgical Treatment Considerations
Medical care and treatment approaches for patients with von Hippel-Lindau (VHL) disease are determined by the specific complications present. Most therapies for complications of VHL disease are surgical (eg, excision of tumors in the CNS, adrenal gland tissue, or kidneys). Patients who undergo nephrectomies due to renal pathology require dialysis or renal transplantation to sustain life.
Belzutifan (Welireg), a hypoxia-inducible factor inhibitor, was approved by the FDA in August 2021. It is indicated for treatment of adults with VHL disease who require therapy for associated RCC, central nervous system hemangioblastomas, or pancreatic neuroendocrine tumors (pNET), not requiring immediate surgery. Belzutifan binds to hypoxia-inducible factor 2 alpha (HIF-2α) and prevents HIF-2α from interacting with hypoxia-inducible factor 1 beta, resulting in reduced transcription and expression of HIF-2α target genes.
Belzutifan approval was based on efficacy results from a phase 2 clinical trial in 61 patients with VHL-associated RCC. Median time from initial radiographic diagnosis of VHL-associated RCC tumors. Time of treatment with belzutifan was 17.9 months. For VHL-associated RCC, median time to response (TTR) was 8 months. For VHL-associated CNS hemangioblastomas, TTR was 3.1 months. For VHL-associated pNET, median TTR was 8.1 months. [22]
Nonsurgical treatment interventions focus on prevention and scheduled surveillance. For example, annual ophthalmologic examination is recommended because of the risk for retinal hemangioblastomas. Early detection and treatment of retinal lesions reduces the risk of visual loss. However, if a patient presents with one manifestation of VHL disease (eg, retinal hemangioblastoma), treating the clinical finding in isolation is not sufficient. The patient is at risk for additional, life-threatening problems that may not be immediately obvious. Thus, the most crucial aspect of medical care for these patients is close surveillance and prompt evaluation with appropriate diagnostic imaging.
Increasing knowledge about the molecular role of VHL proteins (pVHLs) has led to clinical trials of several antiangiogenic drugs designed to reduce or prevent tumorigenesis in VHL disease. Positive progress has been made in the use of antiangiogenic medications to treat renal cell carcinoma (eg, the receptor tyrosine kinase inhibitor sunitinib).
Diet and lifestyle
Dietary guidelines for patients with VHL disease have been recommended by the VHL Family Alliance. These guidelines rely more on expert opinion and common sense than on randomized trials. The VHL Family Alliance encourages patients with VHL disease and at-risk family members to make the following lifestyle changes:
-
Increase consumption of phytochemicals, such as grains, cruciferous and other vegetables, fruits, and spices
-
Decrease consumption of protein from fish, poultry, and meat
-
Limit alcohol intake
-
Avoid tobacco products because of their risk associated with renal cancer
-
Avoid chemicals and industrial toxins known to affect VHL-involved organs
Activity
People with VHL disease should not limit their physical activities, except for postsurgical recuperation. Moderate exercise is beneficial. Avoidance of contact sports is recommended if adrenal or pancreatic lesions are present.
Ophthalmic Surgery
von Hippel-Lindau (VHL) disease usually is a progressive disorder, and ophthalmic care should begin as soon as ocular pathology is diagnosed. Surgical treatment may consist of the following:
-
Argon laser photocoagulation
-
Cryotherapy
-
Fluid drainage
-
Scleral buckling
-
Vitreous surgery
-
Penetrating diathermy and endodiathermy
-
Radiation
Argon laser photocoagulation
Argon laser photocoagulation is effective in treating retinal angiomatosis. Treatment involves the use of a large spot size with low-intensity, long-duration burns being directed at the angioma. Repeated laser treatment is required, except for small tumors. Obliteration of the tumor is confirmed by clinical observation and fluorescein angiography. If the tumor turns yellowish, photocoagulation becomes difficult because of the poor penetration of laser light.
Cryotherapy
Cryotherapy, a repetitive freeze/thaw technique, may be used to treat anterior angiomas and larger posterior angiomas. To minimize the risk of hemorrhage, no more than 2 or 3 freeze/thaw cycles should be performed per session. Multiple sessions generally are needed to arrest tumors. Resolution of the macula edema and improved visual acuity are common results of tumor eradication by cryotherapy.
Scleral buckling and fluid drainage
Scleral buckling and fluid drainage methods may be needed to treat larger tumors or tumors associated with retinal detachment. Angiomas resistant to cryotherapy or tumors involving subretinal exudation also may require these interventions. Large tumors can develop surface membranes and vitreous traction, leading to vitreous hemorrhage or rhegmatogenous retinal detachment. These complications may require treatment by vitreous surgery, endodiathermy, or scleral buckling techniques.
Penetrating diathermy and endodiathermy
Penetrating diathermy under a lamellar scleral bed is an effective treatment of larger angiomas. Endodiathermy may be used, instead of vitreous surgical techniques or scleral buckling procedures, to treat vitreous hemorrhage or retinal detachment.
Radiation
Radiation, applied in various forms, is another field among future directions in the care of retinal angiomatosis.
Renal and Neurologic Surgery
Clear cell renal cell carcinoma is the most significant life-threatening tumor that occurs in patients with von Hippel-Lindau (VHL) disease. Once identified, and with limited tumor involvement, partial nephrectomy or radiofrequency ablation is the preferred treatment approach in an attempt to preserve renal function. Total nephrectomy is reserved for those patients with extensive tumor involvement. Bilateral nephrectomy ultimately may be required because of the presence of multiple primary renal tumors. After this surgical intervention, renal dialysis or renal transplantation is required to sustain life.
CNS hemangioblastomas typically are not malignant; thus, they can be monitored if their size remains stable and they are not inducing focal neurologic signs. However, if these tumors cause neurologic symptoms, neurosurgical excision is required.
Consultations
Once von Hippel-Lindau (VHL) disease is diagnosed, consultation with a medical geneticist and a genetic counselor is imperative. Genetic counseling is critical because the diagnosis of VHL disease affects family members who are at risk. Genetic counselors are trained to coordinate genetic testing and to interpret results of the mutational analysis, as well as, supply resources and referrals for support groups and needed clinical services. Psychological and/or psychiatric intervention may be indicated to address the emotional strain of being diagnosed with a chronic disease.
The multiple-organ nature of this disease necessitates that the following specialists be involved in the patient's care:
-
Consult an ophthalmologist due to the risk of retinal angiomatosis.
-
Obtain consultations with surgeons as needed, depending on the results of surveillance imaging studies as detailed elsewhere in this article; typically, a nephrologist and/or urologist are consulted with the findings of renal masses, and neurosurgeons are consulted concerning CNS masses.
-
An endocrinologist should be consulted to assist in the surveillance and interpretation of laboratory testing for pheochromocytoma.
Long-Term Monitoring
The pleiotropic clinical manifestations of von Hippel-Lindau (VHL) disease and the potential for malignancy require a lifelong strategy of surveillance (particularly for the neurologic, ocular and renal systems) to enable early detection and treatment of complications. A clear plan for scheduled surveillance should be made, with copies of this plan provided to the primary care physician and to the patient (or to the parents/caregivers of pediatric patients).
A medical surveillance strategy for affected patients and at-risk relatives is as follows:
-
Obtain a medical history and perform a physical examination annually, including blood pressure monitoring.
-
Obtain laboratory studies (urine and blood samples) annually, starting at age 2 years; testing consists of urinalysis, urine cytologic examination, 24-hour urinary catecholamine metabolites, CBC count, electrolytes, renal function (BUN, Cr), and plasma catecholamines.
-
Perform annual direct and indirect ophthalmoscopic examinations; this is best if begun at age 2 years.
-
Perform an audiologic examination at the first sign of hearing problems, vertigo, or tinnitus; in one study of VHL gene mutation carriers, more than 90% of radiologically diagnosed ELSTs were associated with abnormal audiometric findings. [23] When abnormalities are found, a T1-weighted MRI of the temporal bone should be performed.
-
Perform annual abdominal ultrasonography, beginning in the teenage years, to look for abnormalities in the kidneys, adrenal glands, and/or pancreas; concerning findings warrant further investigation with either CT scan or MRI.
-
Perform MRI of the brain and spinal cord every 2-3 years, beginning in the teenage years. This recommendation remains controversial within medical circles because CNS tumors without neurological focality are typically not resected.
Patients comply well with completing these monitoring tests at cited intervals, when the physician communicates directly to the patient or the patient’s caregiver. [24] A case manager or a nurse practitioner can be assigned to ensure proper surveillance. Screening can be discontinued for at-risk relatives aged 65 years or older if no abnormalities are found up to this age.
Patient Education
Patients and families can benefit from contacting the VHL Family Alliance(www.vhl.org) at 800-767-4845 (800-767-4VHL) or by email at info@vhl.org.
In addition, the VHL Alliance has established a Clinical Care Program composed of Clinical Care Centers (CCC) and Comprehensive Clinical Care Centers (CCCC) on both a national and international scope. These Care Centers serve as hubs of clinical care for VHL patients. The program allows patients to be seen by specialists familiar with VHL disease. Care Centers provide timely annual screening and ongoing treatment and are a resource of information. Compassion, knowledge, skill, and coordination of care are critical for patients with VHL disease and their families.
As of 2014, Care Centers now distinguish between medical facilities that provide care to pediatric-only, adult-only, and all patients (both adult and pediatric). National and international locations are cited on the VHL Family Alliance Web site. The goals of the VHL Clinical Care Centers are to improve the diagnosis and treatment of VHL disease, to provide coordinated care across various medical specialties, and to provide resource centers for patients, families and physicians.
The National Cancer Institute at the US National Institutes of Health (NIH) sponsor a Web site (www.cancer.gov )with general information useful to patients with VHL disease and their families. Genetic counseling providers are cited on the National Society of Genetic Counselors Web site (www.nsgc.org).
Additional support organizations include the Kidney Cancer Association(www.kidneycancer.org) and the NIH/National Eye Institute (www.nei.nih.gov).
-
von Hippel-Lindau disease. Clinical picture of the retina, showing a pair of dilated and tortuous feeder vessels coursing on the surface of the retina from the optic nerve head toward the angioma (which is not seen in this image because it is in the extreme periphery).
-
von Hippel-Lindau disease. Color Doppler image of the dilated feeder vessels with flow patterns in opposite directions as indicated by red for the artery and blue for the vein. The abbreviation ON designates the optic nerve with its blood vessels.
-
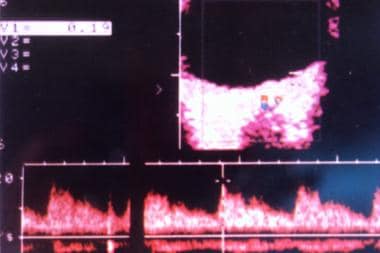
von Hippel-Lindau disease. Spectral display of the aberrant feeder vessels.
-
T1-weighted, transaxial, gadolinium-enhanced magnetic resonance imaging (MRI) scans show a well-defined, hypervascular, enhancing mass.
Tables

What would you like to print?
- Overview
- Physiology
- Prognosis
- Physical Examination
- Differential Diagnosis
- Laboratory Studies
- Imaging Studies
- Evaluation of At-Risk Family Members
- Nonsurgical Treatment Considerations
- Ophthalmic Surgery
- Renal and Neurologic Surgery
- Consultations
- Long-Term Monitoring
- Patient Education
- Show All
- Media Gallery
- References





