Practice Essentials
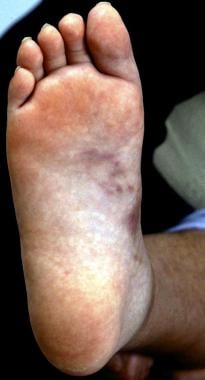
Vasculitis is defined as inflammation of blood vessels. It may result in vessel wall thickening, stenosis, and occlusion with subsequent ischemia. Necrotizing inflammation can completely destroy segments of the wall. Vasculitis can involve vessels of any size and can affect any organ system. The clinical presentation varies according to the histologic type of inflammation, the size of the involved blood vessel segment, and the distribution of the involved vessels. The image below depicts nodules in cutaneous polyarteritis nodosa (PAN), a systemic vasculitis characterized by necrotizing inflammatory lesions that affect medium-sized and small muscular arteries.
Many subtypes of vasculitis are recognized. This article focuses on the primary systemic vasculitides of childhood. The most common forms of systemic vasculitis in children are Henoch-Schonlein purpura (HSP) and Kawasaki disease (KD). Other subtypes of primary childhood vasculitis are much rarer. Vasculitis can also occur secondary to another disease, and this should be ruled out. Diseases associated with secondary vasculitis include infection, malignancy, collagen vascular disease (eg, systemic lupus erythematosus, dermatomyositis), and drug hypersensitivity. See Differentials.
Classification Guidelines
Practitioners may find it helpful to think of primary systemic vasculitides based on the predominant size of the involved vessels, as suggested by the Chapel Hill Classification. [1, 2] See the image below.

However, an overlap of vessel sizes affected within the diseases is noted, and the phenotype and pattern of organ involvement in vasculitis seems to be independent of vessel size. For example, Kawasaki disease is characterized by a mucocutaneous syndrome and coronary artery inflammation, whereas polyarteritis nodosa (PAN), another medium-vessel vasculitis, manifests with nodular skin lesions, neuropathy, and hypertension.
As such, the classification of childhood vasculitides incorporates both vessel size and organ manifestations. This classification was proposed at a consensus conference in 2005 and was endorsed by Paediatric Rheumatology European Society (PRES) and the European League against Rheumatism (EULAR). These criteria represent a modification and adaptation of existing American College of Rheumatology and Chapel Hill criteria for vasculitis in adults.
The EULAR and PRES Classification of Childhood Vasculitis
The predominantly large vessel vasculitis is Takayasu arteritis.
Predominantly medium-sized vessel vasculitis includes the following:
-
Childhood polyarteritis nodosa
-
Cutaneous polyarteritis
-
Kawasaki disease
-
Primary vasculitis of the CNS, angiography-positive primary angiitis of the CNS (PACNS)
Predominantly small vessel vasculitis is divided into granulomatous and nongranulomatous. Granulomatous includes the following:
-
Granulomatosis with polyangiitis (formerly known as Wegener granulomatosis)
-
Churg-Strauss syndrome
Nongranulomatous includes the following:
-
Microscopic polyangiitis
-
Henoch-Schönlein purpura
-
Isolated cutaneous leukocytoclastic vasculitis
-
Hypocomplementemic urticarial vasculitis
-
Primary vasculitis of the CNS, angiography-negative, small vessel PACNS
Other vasculitides includes the following:
-
Behçet disease
-
Antiglomerular basement membrane (GBM) antibody disease
-
Vasculitis secondary to infection (including hepatitis B associated polyarteritis nodosa), malignancies, and drugs, including hypersensitivity vasculitis
-
Vasculitis associated with connective tissue diseases
-
Cogan syndrome
-
Unclassified
Specific Classification Criteria
Large-vessel vasculitides
Note that the main large vessel vasculitis that affects children is Takayasu arteritis, and that temporal arteritis is not seen in the pediatric population. Takayasu arteritis is characterized by transmural inflammation and evidence of intramural giant cells. It involves the aorta and its major branches. Characteristic clinical features are caused by stenotic large vessels and subsequently decreased blood supply to the organ systems. Classically, children present with claudication, absent peripheral pulses, blood pressure abnormalities, strokes, and features of internal organ ischemia. A literature review by Duarte et al found an estimated prevalence of stroke/transient ischemic attack in patients with Takayasu arteritis of almost 16%. [3]
Classification criteria for Takayasu arteritis includes angiographic abnormalities (conventional, CT, or MRI) of the aorta or its main branches (mandatory criterion), plus at least one of the following features:
-
Decreased peripheral artery pulse and/or claudication of extremities
-
Blood pressure difference of more than 10 mm Hg
-
Bruits over aorta and/or its major branches
-
Hypertension (related to childhood normative data)
Medium-vessel vasculitides
The most common childhood medium-vessel vasculitis is Kawasaki disease. It is a necrotizing vasculitis that has a predilection for the coronary arteries. Classic features include prolonged fever, mucocutaneous changes and lymphadenopathy. Other manifestations may include irritability, arthritis, and abdominal pain.
Classification criteria for Kawasaki disease include a fever persisting for at least five days (mandatory criterion) plus 4 of the following 5 features:
-
Changes in peripheral extremities or perineal area
-
Polymorphous exanthema
-
Bilateral conjunctival injection
-
Changes of lips and oral cavity (injection of oral and pharyngeal mucosa)
-
Cervical lymphadenopathy
Childhood polyarteritis nodosa is a necrotizing vasculitis of medium-sized arteries and is recognized in distinct systemic and cutaneous forms.
Systemic polyarteritis nodosa involves all organ systems and the presentation widely varies. [4] Features include vasculitic skin lesions, hypertension, neuropathy, and myalgia. Note that renal involvement does not manifest as glomerulonephritis, as occurs with small-vessel disease. Unlike in adults, angiographic or biopsy evidence of vasculitis is required to make this diagnosis.
Classification criteria for childhood polyarteritis nodosa include a systemic illness characterized by the presence of either a biopsy finding that reveals small and mid-size artery necrotizing vasculitis or angiographic abnormalities (aneurysms or occlusions), plus at least 2 of the following:
-
Skin involvement (eg, livedo reticularis, tender subcutaneous nodules, other vasculitic lesions)
-
Myalgia or muscle tenderness
-
Systemic hypertension, relative to childhood normative data
-
Mononeuropathy or polyneuropathy
-
Abnormal urine analysis and/or impaired renal function (glomerular filtration rate of < 50% normal for age)
-
Testicular pain or tenderness
-
Signs or symptoms suggesting vasculitis of any other major organ system (GI, cardiac, pulmonary, CNS)
Cutaneous polyarteritis nodosa is characterized by the presence of subcutaneous nodular, painful, nonpurpuric lesions with or without livedo reticularis and absence of systemic involvement. However, more than half of patients also have myalgia, arthralgia, and nonerosive arthritis. Cutaneous polyarteritis nodosa has been associated with serological or microbiological evidence of streptococcal infection in 40% of patients. [4]
Childhood PACNS is defined by clinical evidence of a newly-acquired focal or diffuse neurologic deficit plus angiographic or histologic evidence of CNS vasculitis, in the absence of a systemic condition associated with these findings. Two clinically and radiologically distinct types of childhood PACNS are noted: large-medium vessel (angiography-positive) and small vessel (angiography-negative). These have different clinical presentations (see Primary CNS Vasculitis of Childhood).
Small-vessel vasculitides
Henoch-Schönlein purpura is the most common vasculitis in children, and is associated with immunoglobulin A (IgA) immune deposition in small vessels. Presenting features include palpable purpura, abdominal pain (which may be associated with GI hemorrhage and/or intussusception), arthritis, and evidence of glomerulonephritis (hematuria, proteinuria).
Classification criteria for Henoch-Schönlein purpura include palpable purpura (mandatory criterion) in the presence of at least one of following 4 features:
-
Diffuse abdominal pain
-
Any biopsy finding that reveals predominant lgA deposition
-
Arthritis or arthralgia (arthritis is acute in any joint)
-
Renal involvement (any hematuria, proteinuria)
Antineutrophil cytoplasmic antibody (ANCA) positive small-vessel vasculitides are also seen in the pediatric population. Granulomatosis with polyangiitis (GPA) (formerly known as Wegener granulomatosis) is a granulomatous vasculitis that most commonly involves the sinopulmonary system but can involve any organ system. Presenting features may include purpuric rash, recurrent sinusitis, epistaxis, shortness of breath, and/or hemoptysis from alveolar hemorrhage. It is also commonly associated with a necrotizing glomerulonephritis that may cause significant renal impairment. [5] Patients with Wegener granulomatosis have a positive ANCA finding in 90% of cases, most frequently in a cytoplasmic pattern (c-ANCA), with antibodies against proteinase 3 (anti-PR3). [6]
Classification criteria for GPA include 3 of the following 6 features:
-
Abnormal urinalysis findings (hematuria and/or significant proteinuria)
-
Granulomatous inflammation on biopsy (If a kidney biopsy is done it characteristically shows necrotizing pauci-immune glomerulonephritis.)
-
Nasal sinus inflammation
-
Subglottic, tracheal, or endobronchial stenosis
-
Abnormal chest radiography or CT findings
-
PR3 ANCA or c-ANCA staining
Microscopic polyangiitis (MPA) is a necrotizing vasculitis associated with glomerulonephritis and pulmonary capillaritis. Presenting features include purpuric rash, proteinuria and/or hematuria, hemoptysis, CNS involvement, and arthralgias. [7] MPA is associated with ANCA, most commonly with a perinuclear pattern (p-ANCA) and antibodies against myeloperoxidase (anti-MPO).
Churg-Strauss syndrome (CSS) is an eosinophilic granulomatous vasculitis characterized predominantly by pulmonary involvement. Patients typically have a previous history of asthma, allergic rhinitis, and/or sinusitis. A characteristic feature is the finding of nonfixed pulmonary infiltrates. Eosinophilic infiltration results in multiorgan involvement, including neuropathy and cardiovascular disease (pericarditis). [8] ANCA positivity is seen in approximately 40% of patients, usually with an “atypical” or “indeterminate” pattern.
Isolated cutaneous leukocytoclastic vasculitis can be either primary (rarely) or secondary to various medications, infections, or collagen vascular disease.
Hypocomplementemic urticarial vasculitis is a cutaneous vasculitis that may result from primary hypocomplementemia or as part of a disease associated with low complement levels (eg, systemic lupus erythematosus).
Other vasculitides
Behcet disease involves vessels of all sizes. The diagnosis is made clinically in patients with recurrent oral ulcers who also have recurrent genital ulcers, uveitis, various skin lesions, and/or a positive pathergy test.
Anti-GBM antibody disease/Goodpasture syndrome is a type of vasculitis caused by deposition of anti-GBM antibodies in small vessels of lungs and kidneys. Goodpasture syndrome clinically manifests with rapidly progressive glomerulonephritis and/or pulmonary hemorrhage (pulmonary renal syndrome).
Thrombophlebitis refers to inflammation of a vein associated with the formation of a blood clot. This may arise due to an interaction of endothelial injury, stasis of blood, and a hypercoagulable state. Risk factors include intravenous catheter placement, immobilization, malignancy, inherited prothrombotic condition (eg, factor V Leiden, antithrombin III deficiency), antiphospholipid antibody syndrome, and Behçet disease. One study found vascular involvement in 14% of patients with Behçet disease, most commonly manifesting as superficial vein thrombophlebitis or deep vein thrombosis. [9]
Pathophysiology
Vessel inflammation occurs by various mechanisms in this heterogenous group of diseases. The histopathological pattern of inflammation is a characteristic feature of the vasculitis subtypes.
Lymphocytic/giant cell-mediated vasculitis
Takayasu arteritis and temporal arteritis (in adults) both involve large elastic arteries and share a similar histopathology. This form of vasculitis is T-cell dependent, and CD4+ T cells are the main players in the process. Dendritic cells within the arterial adventitia recruit and stimulate the CD4+ cells, which then activate the monocytes and macrophages that mediate oxidative injury of the vessel wall. Vascular lesions are characterized by a panarteritis with mononuclear infiltration of all layers of the arterial wall. Typically, activated T cells and macrophages are arranged in granulomas, and multinucleated giant cells are present. Often, the intimal layer is hyperplastic, leading to concentric occlusion of the lumen. Also, the end stage of giant-cell aortitis may be complicated by the formation and rupture of aneurysms. [10]
Necrotizing vasculitis
Kawasaki disease and polyarteritis nodosa are examples of necrotizing vasculitis. Possible etiologies in the case of Kawasaki disease include infectious agents and/or superantigen-mediated activation of lymphocytes. The inciting factors in polyarteritis nodosa are less well understood; however, in developing countries, it has been associated with hepatitis B or C. Pathologically, segmental transmural inflammation of muscular arteries is noted. Nodule (vascular narrowing) and aneurysm formation result from panmural fibrinoid necrosis. Note that aneurysmal dilatation of the arterial wall is a common feature of necrotizing vasculitis. Typically, immunofluorescence for immunoglobulin or complement deposition is negative. [11]
Antibody-mediated vasculitides
A review by Jennette and Falk discusses the scientific evidence showing that ANCA immunoglobulin Gs (IgGs) are involved in the pathogenesis of small vessel vasculitides such as GPA and MPA. [12]
ANCA antibodies are directed towards cytoplasmic proteins within neutrophils and monocytes (eg, PR3, MPO), which may also be expressed at the cell surface, particularly on stimulated cells. In vitro studies have shown that ANCA IgGs can directly activate neutrophils and monocytes by both Fc receptor engagement and direct Fab2 binding to antigen. These activated cells interact with endothelial cells via adhesion molecules and release inflammatory mediators, such as toxic granule enzymes and reactive oxygen metabolites that cause apoptosis and necrosis. In addition, anti-MPO IgG may activate MPO itself triggering an oxidative burst and resulting in severe endothelial damage. ANCA-activated neutrophils may release factors that activate the alternative complement pathway, which initiates an amplification loop that mediates the severe necrotizing inflammation of ANCA disease. [12]
In vivo studies also support this pathogenesis; for example, injection of mice with anti-MPO antibodies results in the development of necrotizing and crescentic glomerulonephritis and pulmonary capillaritis. [13]
A study that sought to assess clinical and B cell biomarkers to predict relapse after rituximab in antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV) using retreatment on clinical relapse strategy found that naïve B-lymphopenia may be a biomarker of disease activity in AAV. [14, 15]
Antibody deposition
Henoch-Schönlein purpura is generally characterized by the deposition of IgA antibodies in affected tissues. Histopathologically, the typical finding on skin biopsy is leukocytoclastic vasculitis, with perivascular accumulation of neutrophils and mononuclear cells. Immunofluorescence demonstrates IgA, C3, and fibrin in the walls of affected vessels, including the postcapillary venules within the dermis, and the endothelial and mesangial cells of the kidney. Elevated serum IgA and circulating IgA-containing immune complexes may be present in some patients. One study showed that galactose deficiency of O-linked glycans in the hinge region of IgA1 has been associated with Henoch-Schönlein purpura. [16]
In anti-GBM antibody disease, circulating antibodies bind to type IV collagen within the glomerular basement membrane. Immunofluorescence study of renal biopsies demonstrates linear deposition of IgG along the glomerular basement membrane. Pulmonary hemorrhage occurs when these antibodies have access to the alveolar basement membrane.
Etiology
Secondary vasculitis
Infectious causes include the following:
-
Streptococcus
-
Herpes simplex
-
Varicella
-
Epstein-Barr virus (EBV)
-
Cytomegalovirus (CMV)
-
Parvovirus B19
-
Hepatitis B and C
-
Candida albicans
Systemic rheumatic disease causes include the following:
Inflammatory bowel disease is a cause.
Lemierre syndrome is an anaerobic suppurative thrombophlebitis of the internal jugular vein and is most commonly a complication of pharyngeal, dental, or mastoidal infection.
Hypocomplementemic urticarial vasculitis is also a cause.
Malignancy causes include the following:
-
Leukemia
-
Lymphoma
-
Bronchopulmonary blastoma, angioblastoma (both rare)
Hypersensitivity vasculitis (leukocytoclastic vasculitis) and drug-induced ANCA vasculitis (propylthiouracil, hydralazine) are also both noted.
Epidemiology
United States statistics
Henoch-Schönlein purpura is the most common vasculitis in childhood with an incidence of approximately 1 in 5000 children annually. [17]
In North America, Kawasaki disease occurs in about approximately 20 per 100,000 children younger than 5 years. [18]
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is reported to occur in 0.03-3.2 per 100,000 children per year. [19]
The other vasculitides are quite rare in childhood.
International statistics
In Japan, the incidence of Kawasaki disease is 188 per 100,000 children per year. [18]
The global incidence of Henoch-Schönlein purpura is 10-20 cases per 100,000 children per year. [20]
Race-, sex-, and age-related demographics
Race
The vasculitides are seen in patients of all races and ethnicities, but some notable patterns of distribution are noted.
Kawasaki disease is most common in children of Japanese and other Asian descent.
Henoch-Schönlein purpura is more common in Whites.
Takayasu arteritis is more common in the Asian population.
Behçet disease is more common in Turkey, the Middle East and eastern Asia.
Sex
Henoch-Schönlein purpura has a male-to-female ratio of 2:1.
Kawasaki disease has a male-to-female ratio of 1.6:1.
Polyarteritis nodosa has a slight male preponderance.
Takayasu arteritis has a strong female preponderance.
Age
Henoch-Schönlein purpura has a peak age of onset at 3-10 years; 75% of patients are younger than 10 years.
Kawasaki disease has a mean age of onset 4.3 years; 80% of patients are younger than 5 years. [17]
Polyarteritis nodosa has a peak age of onset at 9-11 years.
Takayasu arteritis most commonly presents in the second and third decades of life; 20% are younger than 20 years.
Prognosis
Prognosis is related to the degree of end-organ involvement. Generally, ANCA-associated vasculitis is associated with a poorer prognosis
Recurrence rate in Kawasaki disease is approximately 2%. Patients with Kawasaki disease and large coronary aneurysms are at risk for multiple complications, including stenosis and obstruction, myocardial infarction, and dysrhythmias. Some experience with bypass grafting for revascularization has been reported very good success. [21, 22] In 2014, the largest US study of longer-term cardiac outcomes after Kawasaki disease reported a low rate of adverse cardiovascular events through age 21 years. [23]
The recurrence rate in Henoch-Schönlein purpura is approximately 30%.
A cohort study by Batu et al found that fever and renal involvement were more common in patients with COVID-19–associated pediatric IgA vasculitis/Henoch-Schönlein purpura than in those who developed pediatric IgA vasculitis/Henoch-Schönlein purpura before the pandemic. In addition, patients with COVID‐19–associated vasculitis were less likely to recover without treatment or to achieve a complete recovery. Patients with multisystem inflammatory syndrome in children (MIS-C) were excluded from the study. [24]
Morbidity/mortality
Morbidity and mortality in systemic vasculitides has been reviewed by Phillip and Luqmani; however, it is mainly based on adult data. [25]
In Kawasaki disease, acute mortality is 0.12% (mainly cardiac-related deaths). With appropriate treatment, the rate of coronary aneurysm development is approximately 2-4%.
Henoch-Schönlein purpura is usually a self-limited condition. The long-term prognosis relies mainly on the severity of renal involvement. The overall risk of end-stage renal disease is 1-3%, but this risk can increase to 20% if the patient presents with significant nephritis or nephrotic syndrome. [26] In adults with Henoch-Schönlein purpura, a higher rate of renal impairment is noted, and the 5-year survival rate is only approximately 75%.
Takaysu arteritis is associated with a 10-year survival rate of approximately 87%.
Polyarteritis nodosa and Churg-Strauss syndrome are associated with a 5-year survival rate of 75-80%.
Microscopic polyangiitis is associated with a 5 year survival rate of 45-75%.
Granulomatosis with polyangiitis (formerly Wegener granulomatosis) is associated with significant morbidity and mortality. Approximately 11% of patients require mechanical ventilation and/or require dialysis. [19] Survival is approximately 75% at 5 years in adults; survival data are not reported in pediatric patients.
Long-term data in childhood PACNS are lacking, but early recognition and treatment have been associated with good recovery. [27]
Complications
Complications may include the following:
-
Destruction of paranasal sinuses
-
Subglottic stenosis requiring tracheostomy
-
Life-threatening pulmonary hemorrhage
-
Renal insufficiency requiring dialysis or transplant
-
Digital gangrene with autoamputation
-
Stroke
-
Myocardial infarction
-
Sepsis
-
Morbidity associated with immunosuppressive medications
-
Death
Patient Education
Patients receiving corticosteroids should be advised of possible side effects, including weight gain, sleep disturbance, hirsutism, glucose intolerance, and hypertension.
Patients receiving immunosuppressive agents should be instructed to seek medical attention with any sign of infection.
Patients taking methotrexate should avoid alcohol and other hepatotoxic substances.
Discussion of adequate birth control measures with patients of childbearing age is necessary if they are treated with teratogenic medications (eg, methotrexate, warfarin).
-
Preferred sites of vascular involvement by selected vasculitides.
-
Patient with Wegener granulomatosis and saddle-nose deformity.
-
Tender erythematous nodules in cutaneous polyarteritis nodosa (PAN).
-
Nodules on sole of foot in cutaneous polyarteritis nodosa (PAN).
-
Necrotic lesions of polyarteritis nodosa (PAN).
-
Chest radiography in Churg-Strauss syndrome (CSS) with pulmonary infiltrates.
-
CT of sinuses in a patient with Wegener granulomatosis (WG) showing erosion and loss of sinus walls.
-
CT chest in a patient with Churg-Strauss syndrome (CSS) showing multiple nodules.
Tables
Category |
Definition |
Localized |
Upper and/or lower respiratory tract disease without any other systemic involvement or constitutional symptoms |
Early systemic |
Any, without organ-threatening or life-threatening disease |
Generalized |
Renal or other organ-threatening disease, serum creatinine >500 μmol/L (5.6 mg/dL) |
Severe |
Renal or other vital organ failure, serum creatinine >500 μmol/L (5.6 mg/dL) |
Refractory |
Progressive disease unresponsive to glucocorticoids and cyclophosphamide |







