Practice Essentials
Immunoglobulin A (IgA) nephropathy (also called Berger disease) is a primary glomerulonephritis that is found across the globe. It was first described by Berger and Hinglais in 1968, based on the finding of predominant IgA deposition in the mesangium with a mesangial proliferation. The clinical spectrum varies from asymptomatic microhematuria to rapidly progressive glomerulonephritis. The vast majority of patients are characterized by recurrent episodes of gross hematuria, which usually occurs in concomitance with mucosal infections of the upper respiratory tract or other infections, or by asymptomatic microscopic hematuria with or without proteinuria. [1]
Although it can present at any time, the peak incidence of disease is in the second and third decades of life. A male-to-female ratio of 2:1 is observed in North American and Western European populations, although this difference is not observed among populations in the Pacific.
IgA nephropathy occurs with greatest frequency in Asians and Whites and is relatively rare in Blacks. In a Chinese study, IgA nephropathy constituted 45% of all cases of primary glomerulonephritis. [2] However, IgA deposits may also be seen on kidney biopsy findings in individuals with no evidence of renal disease. The reported incidence rate of mesangial IgA deposition in apparently healthy individuals is 3-16%. These cases had no clinical features of nephritis, but their renal biopsy findings were consistent with IgA nephropathy.
Spontaneous remission has been reported in children and adults. Secondary IgA nephropathy is also associated with various underlying disease processes. It was initially considered a benign condition, but extended follow-up indicates that IgA nephropathy does lead to significant kidney damage, and progressive disease develops in 20-30% of children 15-20 years after disease onset. Advanced age, hypertension, proteinuria, and impaired renal function at presentation are poor prognostic indicators.
Pathophysiology
IgA nephropathy, the most common form of primary glomerulonephritis worldwide, is defined by predominant IgA1 deposits in the glomerular mesangium (see the image below). Over the last decade, the understanding of the pathogenesis of IgA nephropathy has improved. It is believed that IgA nephropathy represents abnormal polyclonal IgA production, specifically a post-translational glycosylation defect. This abnormal glycosylation impairs the normal clearance from the bloodstream of the circulating IgA molecules, as well as predisposing their deposition within the kidneys. [3]
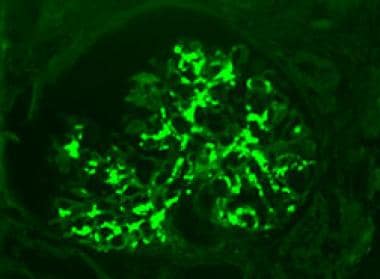 Mesangial deposits of immunoglobulin A (IgA). Fluoresceinated Anti-IgA Antibody, Immunofluorescence microscopy, original magnification 400x. Image courtesy of Patrick D Walker, MD.
Mesangial deposits of immunoglobulin A (IgA). Fluoresceinated Anti-IgA Antibody, Immunofluorescence microscopy, original magnification 400x. Image courtesy of Patrick D Walker, MD.
Immunoglobulin antibody A is a major serum immunoglobulin and the predominant antibody class in the external secretions that bathe mucosal surfaces. This plays a key role in immune protection. Indeed, the body expends considerable energy in producing IgA, such that the daily production of IgA exceeds that of all the other antibody classes combined.
IgA, at concentrations of about 2-3 mg/mL, is the second most prevalent antibody in serum after IgG, which is normally present at about 12 mg/mL. Because serum IgA is metabolized 5 times faster than IgG, the production rates of serum IgA and IgG must be similar. Serum IgA is predominantly monomeric in nature; the secretory IgA (S-IgA) is chiefly polymeric, comprising mainly dimeric forms of IgA containing the J (joining) chain. S-IgA serves various functions to protect the vast surface area (approximately 400 m2) occupied by mucosal surfaces, such as the linings of the respiratory, GI, and genitourinary tracts. As the major class of antibody present at these sites, S-IgA can be considered an important first line of defense against many invading pathogens.
Among abnormalities of the IgA immune system reported so far in IgA nephropathy, aberrant O-linked glycosylation in the hinge region of IgA1 is the most consistent finding. IgA1 molecules bearing abnormal glycosylation have been found in serum, tonsillar lymphocytes, and elute from mesangial deposits, and they are characterized by decreased O-linked N -acetylgalactosamine residues with or without alteration in the terminal sialylation of the O-linked sugars. [4]
Increasing evidence supports the underglycosylated IgA-containing immune complex including IgG antibodies against the glycans of the hinge region of IgA1 are key factors for mesangial deposition and then trigger inflammation and glomerular injury. The polymeric IgA is produced after an aberrant mucosal IgA response. The displacement of mucosal B cells to systemic lymphoid organs and bone marrow may arise from abnormal trafficking of lymphocytes along the mucosa–bone marrow axis, involving changes of chemokines and adhesion molecules. [5]
Galactose-deficient IgA1 is recognized by unique autoantibodies, resulting in the formation of pathogenic immune complexes that ultimately induce glomerular injury. Thus, formation of the galactose-deficient IgA1-containing immune complexes is a critical factor in the pathogenesis of IgA nephropathy. [3]
Current data indicate that at least 4 processes contribute to development of IgA nephropathy. Patients with IgA nephropathy often have a genetically determined increase in circulating levels of IgA1, with galactose-deficient O-glycans in the hinge-region (Hit 1). This glycosylation aberrancy is, however, not sufficient to induce renal injury. Synthesis and binding of antibodies directed against galactose-deficient IgA1 are required for the formation of immune complexes that accumulate in the glomerular mesangium (Hits 2 and 3). These immune complexes activate mesangial cells, inducing proliferation and secretion of extracellular matrix, cytokines, and chemokines, which results in renal injury (Hit 4). [6]
The galactose-deficient IgA, through interaction with RR Fc alpha/gamma, may activate circulating lymphocytes and monocytes and enhance their response to chemoattractants produced by the mesangial cell, thus causing the inflammatory infiltrate to initiate and maintain the interstitial injury.
Aberrant O-galactosylation has been found in serum IgA1, in IgA1 isolated from tonsillar lymphocytes, and in IgA1 eluted from mesangial deposits. Evidence suggests that changes in IgA1 O-galactosylation lead to IgA immune complex formation and mesangial IgA deposition. Mesangial cells exposed to these IgA immune complexes proliferate and adopt a proinflammatory phenotype; they secrete cytokines, chemokines, growth factors, and extracellular matrix components, promoting glomerular inflammation and glomerulosclerosis.
Recent evidence suggests that the control of IgA1 O-glycosylation is linked to class switching from IgD to IgA1 synthesis and that the pattern of IgA1 O-glycosylation may be programmed at the time of initial antigen encounter. IgA1 glycosylation varies between systemic and mucosal sites, and the association of aberrant IgA1 galactosylation with low-affinity, polymeric IgA1 antibodies against mucosal antigens suggests undergalactosylated IgA1 may, in fact, be a mucosal glycoform of IgA1. Although suited to the mucosal compartment, when these IgA1 glycoforms enter the systemic circulation in appreciable quantities, they deposit in the mesangium and trigger glomerular inflammation. [7]
Clinical onset is frequently heralded by synpharyngitic hematuria, macroscopic hematuria during an upper respiratory tract infection. Clinical and laboratory data support a postulated extrarenal origin of the glomerular IgA1, likely derived from circulating immune complexes containing polymeric IgA1, deficient in galactose in the hinge-region O-glycans, bound by antiglycan antibodies. The galactose deficiency affects IgA1 induced by mucosal antigens and elevated circulating levels of this abnormal IgA1 are hereditable, suggesting interactions of genetic and environmental factors.
An abnormal mucosal immune response resulting in production of galactose-deficient IgA1 in IgA nephropathy patients is supported by several observations: the aberrant glycosylation affects mostly polymeric IgA1 produced by mucosal-associated IgA1-secreting cells (including those from tonsils), the synpharyngitic nature of the macroscopic hematuria, and the association of disease severity with polymorphisms of a pattern-recognition receptor, TLR9. [8]
Recurrence of IgA nephropathy has been reported in allograft, and rapid disappearance of IgA deposits is observed when kidneys with IgA deposits are transplanted in a patient without IgA nephropathy.
In the next few years, advances recently added to the knowledge of the pathogenesis of nephropathy IgA1 could provide new variables that may allow classification of patients based not only on clinical and morphological criteria but also on a greater understanding of the pathogenic basis.
IgA nephropathy and Henoch-Schonlein purpura (HSP)
IgA nephropathy and HSP share many morphologic and immunopathologic features. The most striking similarities between IgA nephropathy and HSP nephropathy (HSN) are mesangial IgA deposition, elevated serum IgA level, and IgA circulating immune complexes. The glomerular changes (diffuse or focal mesangial proliferation) in HSN are essentially the same as those in IgA nephropathy. An infective episode precedes HSN in 30-50% of patients, and the presence of Haemophilus influenzae antigen in the glomerular mesangium and the presence of IgA antibody against H influenzae in sera have been reported in patients with HSN.
Both have broadly similar geographic distributions and are rare in Black persons. Coexistence in different members of the same family has been reported. Both can be encountered consecutively in the same patient. They have been described in twins, and they bear identical pathological and biological abnormalities. Kidney biopsy does not reveal any differences between the 2 conditions.
Despite these similarities, the 2 conditions are clinically different, and the pathogenesis is not clear. HSP is an acute condition, with a glomerular lesion, and mostly nonprogressive after the onset. Meanwhile, IgA nephropathy is a chronic progressive lesion, which may eventually lead to renal failure. IgA nephropathy has a male predominance. HSP occurs mostly in young children and is rare in adults, whereas IgA nephropathy mainly occurs in older children and young adults. The following are a number of useful distinctive features:
-
The peak age ranges from 15-30 years for a diagnosis of IgA nephropathy, whereas HSP is mainly seen in childhood
-
The extrarenal manifestations of HSP (skin, GI, CNS, joints) are not seen in IgA nephropathy
-
HSP has been described in association with hypersensitivity
-
Endocapillary and extracapillary inflammations, as well as fibrin deposits in the glomerulus, are more frequent in HSP
-
No major biological differences have been found between the 2 illnesses, except for a larger size of circulating IgA-containing complexes (and a greater incidence of increased plasma IgE levels in HSP
-
As tissue infiltration by leukocytes is a major feature of HSP vasculitis, a possible role of a more potent activation of the latter cells by IgA-containing complexes and/or circulating chemokines in HSP can be considered
Oxford classification of IgA nephropathy
A Working Group of the International IgA Nephropathy Network and the Renal Pathology Society have developed a consensus on the pathologic classification of IgA nephropathy. [9] The goal of the new classification was to identify specific pathological features that more accurately predict risk of progression of renal disease in IgA nephropathy. Clinical data and adequate renal biopsy material from 265 patients with IgA nephropathy were collected from 8 countries on 4 continents. Five centers from Asia, 6 from Europe, 2 from United States, 1 from South America, and 2 multicenter networks (Canada and the United States) participated in the study. The proportion of children was similar in each continent (approximately 30%). These patients were followed for a median of 5 years.
Several pathologists identified histologic variables by repeated analysis of biopsies that were consistently interpreted with a high degree of reproducibility. The following variables were identified that correlated with renal outcomes (see the image below):
-
Mesangial hypercellularity (M)
-
Endocapillary hypercellularity (E)
-
Segmental glomerulosclerosis (S)
-
Tubular atrophy/interstitial fibrosis (T)
-
Crescents (C)
MEST-C scores have been increasingly used in clinical practice. Combining the MEST score with clinical data at biopsy provides the same predictive power as monitoring clinical data for 2 years. [10]
Mesangial hypercellularity – Mesangial cells are counted per mesangial area, and a score of zero to three is assigned for each glomerulus. A score of zero indicates that fewer than four mesangial cells are present per mesangial area; a score of one indicates that four to five mesangial cells are present per mesangial area; a score of two indicates that six to seven mesangial cells are present per mesangial area; a score of three indicates that greater than eight mesangial cells are present per mesangial area. Scores obtained for all glomeruli are averaged, and the resulting assigned hypercellularity score is either M0 if the mean score is less than 0.5 or M1 if the mean score is greater than 0.5.
Endocapillary hypercellularity – This is defined as present (E1) if hypercellularity is present within glomerular capillary lumens and results in narrowing of the lumens, or absent (E0) if no hypercellularity is present within lumens.
Segmental glomerulosclerosis – This is defined as present (S1) if any part of the glomerular tuft is involved in sclerosis, or absent (S0) if no segmental glomerulosclerosis is present.
Tubular atrophy/interstitial fibrosis – The percentage of the cortical area involved by tubular atrophy or interstitial fibrosis is quantitated. A score of T0, T1, or T2 is given if the percentage of involved cortical area is 0-25%, 26-50%, or >50%, respectively. Note that scoring of tubular atrophy/interstitial fibrosis was preferred to global glomerulosclerosis, as its quantification is less susceptible to error due to subcapsular sampling or paucity of glomeruli on the biopsy.
Crescents – This feature is defined as present if cellular and/or fibrocellular crescents are present in at least one glomerulus (C1), present in at least 25% of glomeruli (C2), or absent (C0). Fibrous crescents are not counted toward this score.
Biopsies with fewer than 8 glomeruli should be considered of uncertain value for prognosis.
Etiology
The cause of primary IgA nephropathy is unknown. The conditions producing secondary mesangial IgA deposition include the following:
-
HSP
-
Chronic ulcerative colitis
-
Dermatitis herpetiformis
-
Psoriasis [11]
-
Lung cancer
-
Colon cancer
-
Monoclonal IgA gammopathy
-
Pancreatic cancer
-
Cirrhosis
-
Pulmonary hemosiderosis
-
Cryoglobulinemia
-
Systemic lupus erythematosus
-
Rheumatoid arthritis
Epidemiology
United States statistics
IgA nephropathy accounts for 5-10% of all primary glomerular diseases occurring in the United States. The prevalence of IgA nephropathy in the general population has been estimated to be about 25-50 cases per 100,000 population. Almost 5% of all biopsied patients have at least some IgA deposits in their glomeruli. The incidence of end-stage renal disease (ESRD) due to IgA nephropathy was 5.5 cases per million population per year; about 8.4 cases for males and 2.7 cases for females.
International statistics
IgA nephropathy has been diagnosed worldwide, but its prevalence in different countries varies. In Pacific countries, particularly in Japan, it accounts for approximately 50% of all primary glomerular diseases. In Europe, it is responsible for 20-30%. The explanation of this apparent variability is uncertain but may be related, in part, to differing indications for renal biopsy in different centers. High incidence rates are reported in Asia, France, Italy, Finland, and southern Europe. Genetic and environmental factors may contribute to geographic differences in prevalence. Population studies in Germany and France have calculated an incidence of 2 cases per 10,000, although autopsy studies performed in Singapore suggest that 2-4.8% of the population may have IgA deposition in their glomeruli.
In the United Kingdom, Canada, and the United States, it is common practice not to recommend renal biopsy for patients presenting with isolated hematuria or mild proteinuria; examination of renal tissue is reserved for those in whom increasing proteinuria or worsening renal function develops. Such reluctance to perform biopsies inevitably reduces the number of cases of IgA nephropathy reported in the general populations of these countries.
Race-, sex-, and age-related demographics
The distribution of IgA nephropathy varies in different geographic regions throughout the world. It is the most common form of primary glomerular disease in Asia, accounting for as much as 30-40% of all biopsy findings, for 20% of biopsies in Europe, and for 10% of all biopsies performed for glomerular disease in North America. The reason for this wide variance in incidence is partly attributable to indications for renal biopsy in Asia compared to those in North America. In the United States, incidence of IgA nephropathy is increased in children who are Asian or White; incidence is lowest in children who are Black.
Incidence is higher in males than in females. Male-to-female ratios of 2:1 and 6:1 have been reported.
IgA nephropathy occurs in persons of all ages but is still most common in the second and third decades of life and is much more common in males than females. IgA nephropathy is uncommon in children younger than 10 years. In fact, 80% of patients are between the ages of 16-35 years at the time of renal biopsy.
Prognosis
Patients with IgA nephropathy who have little or no proteinuria (less than 500 mg/day) have a low risk of progression, at least in the short term. However, progressive proteinuria and kidney function impairment develop in a substantial proportion of patients over the long term. [12, 13] Among patients who develop overt proteinuria and/or elevated serum creatinine concentration, progression to end-stage kidney disease (ESKD) is approximately 15-25% at 10 years and 20-30% at 20 years. [14, 15]
The rate of progression is typically slow, with the glomerular filtration rate (GFR) often falling by as little as 1 to 3 mL/min per year, a change not associated with an elevation in the serum creatinine concentration in the short term. Thus, a stable and normal serum creatinine concentration does not necessarily indicate stable disease. The frequency with which this occurs has been evaluated in studies in which repeat kidney biopsy was used to assess the frequency of progressive disease. [16] In one report, repeat kidney biopsies were performed at 5 years in 73 patients with persistent proteinuria and a normal or near-normal initial serum creatinine concentration. Histologic improvement occurred in only 4%, with 41% remaining stable and 55% showing progressive glomerular and secondary vascular and tubulointerstitial injury. An increase in serum creatinine to more than 1.5 mg/dL (133 micromol/L) was associated with major pathologic lesions. [17]
IgA nephropathy was initially thought to be a benign disease, but it is now recognized that over 25 years of observation, it can slowly progress to end-stage renal disease in up to 50% of affected patients. [18] The remaining patients may sustain clinical remission or have persistent low-grade hematuria and/or proteinuria. The prognosis is difficult to predict with accuracy in an individual patient, but important risk factors for progressive renal disease have been identified.
The patients who develop progressive disease typically have one or more of the following clinical or laboratory findings at diagnosis, each of which is a marker for more severe disease.
-
A reduction in GFR, as manifested by an elevated serum creatinine concentration at diagnosis or during the course of the disease, is associated with a worse renal prognosis [19]
-
Hypertension (BP >95 percentile for height and sex) is predictive of a worse outcome; a higher mean arterial pressure is associated with a higher risk of progressive renal disease [13]
-
With regard to proteinuria, the rate of progression is very low among patients excreting less than 15 mg/kg/d and is greatest among those excreting more than 50 mg/kg/d [20]
Morbidity/mortality
Although IgA nephropathy was thought to carry a relatively benign prognosis, an estimated 1-2% of all patients with IgA nephropathy develop end-stage renal failure each year from the time of diagnosis. In a study of 1900 patients derived from 11 separate series, the long-term renal survival was estimated to be 78-87% within a decade of presentation. Similarly, European studies have suggested that renal insufficiency may occur in 20-30% of patients within 2 decades of the original presentation.
In a study from Hong Kong, patients with mild IgA nephropathy were prospectively followed. [21] Significant proteinuria or renal insufficiency was found in numerous patients, suggesting that a significant risk of progression is present, even in patients who present with milder forms of disease.
Several studies have assessed features that predict a poor prognosis. Sustained hypertension, persistent proteinuria (especially proteinuria >1 g), impaired renal function, and the nephrotic syndrome constitute poor prognostic markers.
Typically, mortality associated with IgA nephropathy is secondary to renal failure or its complications. Morbidity may be subsequent to hypertension, electrolyte abnormalities, or other consequences of reduced renal function.
Familial IgA nephropathy has an increased risk of end-stage renal disease.
Complications
IgA nephropathy can be associated with two types of complications, as follows:
-
First, due to progression of the disease that can lead to CKD-related complications, such as anemia, uremia, secondary hyperparathyroidism, hypertension, bone and mineral disease, growth and development problems, acidosis, and psychosocial issues
-
Second, complications associated with toxicity from the drugs used to control underlying immune disease, such as corticosteroids, mycophenolate mofetil, tacrolimus, and other medications
Primary complications include those related to uncontrolled hypertension (eg, seizure, stroke, end-organ damage), renal insufficiency (eg, growth failure, bone demineralization, anemia), and adverse reactions to one of the prescribed medications.
Patient Education
Inform patients about specific disease processes when possible.
Encourage patients to avoid risk factors, such as smoking, drugs, obesity, and poor medication compliance.
For patient education resources, see Kidney Disease, Urinalysis, and Blood in the Urine.
-
Glomerulus with mesangial hypercellularity and intact capillary loops. Trichrome Stain, original magnification 400x. Image courtesy of Patrick D Walker, MD.
-
Mesangial deposits of immunoglobulin A (IgA). Fluoresceinated Anti-IgA Antibody, Immunofluorescence microscopy, original magnification 400x. Image courtesy of Patrick D Walker, MD.
-
Electron photomicrograph showing mesangial electron dense deposits (arrow). Uranyl acetate and lead citrate stain, original magnification 12,000x. Image courtesy of Patrick D Walker, MD.





