Practice Essentials
Thanatophoric dysplasia (TD) is the most common form of skeletal dysplasia known to be lethal in the neonatal period. The term thanatophoric derives from the Greek word thanatophorus, which means "death bringing" or "death bearing." Salient phenotypic features of TD include macrocephaly, narrow bell-shaped thorax with shortened ribs, normal trunk length, and severe shortening of the limbs. See the image below.
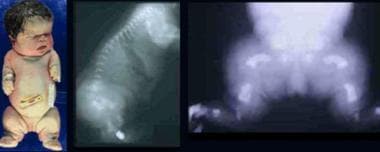 Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).
Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).
TD is divided into 2 clinically defined subtypes: TD type I (TD-I or TD-1) and TD type II (TD-II or TD-2). The clinical subtypes of TD are defined by either a curved or straight appearance of the long bones. TD-I, the more common subtype, is characterized by a normal-shaped skull and curved long bones (shaped like old-fashioned telephone receivers); the femurs are most affected in TD-I. TD-II is associated with a cloverleaf-shaped skull and straight femurs. However, reported cases have cited clinical overlap between these subtypes.
Both TD-I and TD-II are part of a group of skeletal disorders associated with mutations within the Fibroblast Growth Factor Receptor 3 gene (FGFR3). TD-I and TD-II are due to an autosomal dominant point mutation, with the gene responsible, FGFR3, being mapped to the short arm of chromosome 4 (4p16.3). Penetrance of this mutation is 100%. Currently, all cases of TD are due to de novo mutations in FGFR3. Germline mosaicism has not been clearly documented but remains a theoretical possibility. [1, 2]
Signs of thanatophoric dysplasia
Salient phenotypic features in an affected newborn include the following:
-
Severe growth deficiency with an average length of 40 cm (about 16 in) at term
-
Generalized hypotonia
-
Macrocephalic head with frontal bossing and large anterior fontanel
-
Cloverleaf-shaped skull due to premature closure of the cranial sutures
-
Flat facies with low nasal bridge and proptotic eyes
-
Narrow, bell-shaped thorax with short ribs
-
Normal trunk length
-
Protuberant abdomen
-
Micromelia (marked bilateral shortening of the limbs) with redundant skin folds
-
Brachydactyly with a trident hand configuration
Workup in thanatophoric dysplasia
Laboratory studies
The following studies are indicated in suspected cases of TD:
-
Chromosome analysis (karyotype) - To determine the presence or absence of chromosomal abnormalities
-
DNA molecular testing for FGFR3 - Using targeted and sequence mutation analyses
Imaging studies
During the second or third semester of pregnancy, prenatal ultrasonography and computed tomography (CT) scan assessments may be diagnostically helpful.
Postnatal radiography and other imaging studies (CT scanning, magnetic resonance imaging [MRI]) may reveal the following:
-
Enlarged skull and a small foramen magnum, with potential evidence of brain stem compression
-
Central nervous system (CNS) abnormalities such as hydrocephalus, brain stem hypoplasia, temporal lobe malformations, and neuronal migration abnormalities
-
Flattened vertebral bodies (platyspondyly) with wide intervertebral spacing
-
Rhizomelic shortening and irregular metaphyses of the long bones and "telephone receiver–shaped" bowed femurs
Histologic findings
Histologic evaluation of long bone structure in thanatophoric dysplasia shows disruption of endochondral ossification but not of periosteal ossification.
Management of thanatophoric dysplasia
Inpatient care is necessary for newborns diagnosed with thanatophoric dysplasia. If intubation is performed to treat respiratory distress, admission to a neonatal intensive care unit (NICU) is required. If treatment intervention is deferred, palliative care is essential to keep the infant warm, comfortable, and nourished.
Pathophysiology
FGFR3 is part of the tyrosine kinase receptor family. Normally, FGFR3 is a negative regulator of bone growth. Point mutations within FGFR3 causing thanatophoric dysplasia (TD) initiate a gain in function by sending negative signals to the cartilage cells (chondrocytes). These signals occur when ligand binding within the chondrocytes induces receptor homodimerization and heterodimerization. Subsequently, activation of tyrosine kinase function potentiates many effects on cell growth and differentiation.
Researchers believe that mutations in FGFR3 lead to the formation of cysteine residues that create disulfide bonds between extracellular domains of mutant monomers. Activation of the homodimer receptor complex increases its stability and promotes translocation of the complex into the nucleus, where it may interfere with terminal chondrocyte differentiation. Hence, generalized disorganization of endochondral ossification at the bone growth plate occurs.
A study by Martin et al suggested another mechanism for FGFR3 interference with the growth plate. The investigators found evidence that constitutively active FGFR3 causes function loss in chondrocytic primary cilia by impacting cilia length, as well as by impeding the sorting of intraflagellar transport protein 20 and its trafficking to the cilia. [3]
TD-I is caused by several different mutations that affect either the extracellular or intracellular domains of FGFR3. [4] Two missense mutations, R248C and Y373C, account for about 80% of TD-I cases. The more common of these two TD-I point mutations, R248C (known as p.Arg248Cys), is a C→T pyrimidine nucleotide transition and impacts the extracellular domain of FGFR3.
To date, all patients with TD-II have a single point mutation, K650E (known as p.Lys650Glu), with an A→G purine nucleotide transition in the tyrosine kinase domain of FGFR3.
Epidemiology
Frequency
United States
Thanatophoric dysplasia (TD) has an incidence of 1 per 20,000 to 1 per 50,000 births.
International
Incidence in Spain is reported as 1 per 37,000 births.
Mortality/Morbidity
Newborns with TD are frequently stillborn or die shortly after birth. Death can occur within 48 hours and is due to severe respiratory insufficiency from a reduced thoracic capacity and hypoplastic lungs and/or respiratory failure due to brain stem compression.
However, survival beyond the neonatal period is possible in TD. [5, 6] A 2020 report by Carroll et al described a boy aged 9 years with TD-I, while a 2013 article discussed a patient in her late 20s. [7, 8] A study by Ushioda et al found 20 individuals with TD-I in Japan who had survived more than 1 year (age range 1.2-27.8 years). [9]
Sex
Males and females are equally affected.
-
Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).








