Practice Essentials
Smith-Lemli-Opitz syndrome (SLOS) is a multiple congenital anomaly (MCA)/intellectual disability (ID) syndrome caused by a defect in cholesterol synthesis. [1] An autosomal recessive genetic condition, it results from a deficiency of the enzyme 3 beta-hydroxysterol-delta 7-reductase (7-dehydrocholesterol-delta 7-reductase [DHCR7] EC 1.3.1.21), the final enzyme in the sterol synthetic pathway that converts 7-dehydrocholesterol (7DHC) to cholesterol. Smith-Lemli-Opitz syndrome is usually suspected clinically, but the diagnosis must be confirmed by biochemical and/or molecular genetic studies. Currently, no treatment has proven effective long-term for patients with the syndrome. [2]
Affected individuals usually have low plasma cholesterol levels and invariably have elevated levels of cholesterol precursors, including 7DHC. The most severely affected individuals (those with the condition formerly referred to as Smith-Lemli-Opitz syndrome type II) have multiple congenital malformations and are often miscarried or stillborn or die in the first weeks of life. Dysmorphic facial features, microcephaly, second-toe and third-toe syndactyly, other malformations, and intellectual disability (ID) are typical. Mildly affected individuals may have only subtle dysmorphic features and, often, learning and behavioral disabilities.
See the image below.
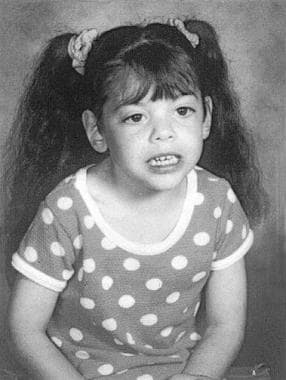 Child with Smith-Lemli-Opitz syndrome.
Child with Smith-Lemli-Opitz syndrome.
Signs and symptoms
The following signs and symptoms may be noted in Smith-Lemli-Opitz syndrome:
-
Lethargy
-
Obtundation or coma
-
Respiratory failure
-
Hearing loss
-
Visual loss
-
Vomiting
-
Feeding difficulties
-
Failure to thrive
-
Constipation
-
Cyanosis
-
Congestive heart failure
-
Photosensitivity
Neuropsychiatric and neurodevelopmental abnormalities are common and include variable intellectual disability (ID), aberrant behavior, and autism.
Workup
Fetal ultrasonography may reveal anomalies suggestive of Smith-Lemli-Opitz syndrome. Confirmatory prenatal diagnostic testing is currently available by genetic mutation analysis.
Postnatally, the syndrome is usually suspected clinically, but biochemical and/or genetic testing is necessary for diagnosis. Plasma total cholesterol and/or low-density lipoprotein (LDL) cholesterol levels may be low but are not universally low. Measurement of plasma sterols, including, at a minimum, cholesterol and 7DHC, is the biochemical test for Smith-Lemli-Opitz syndrome.
Management
As mentioned, no treatment has so far proven effective long-term for patients with Smith-Lemli-Opitz syndrome. [2] Potentially, cholesterol supplementation is a logical treatment because it may be expected to raise plasma and tissue cholesterol levels. By feedback inhibition of hydroxymethylglutaryl-coenzyme-A-reductase, cholesterol supplementation may reduce levels of 7DHC and related cholesterol intermediates that may be toxic. Some suggestion of a potential therapeutic response has been seen with hydroxymethylglutaryl-coenzyme-A-reductase inhibitors (statins), but adverse events are not infrequent. [3]
Pathophysiology
The classic paradigm for the pathogenesis of an inborn error of metabolism includes the accumulation of a toxic precursor and/or deficiency of an essential product as a result of an enzyme deficiency. In the case of Smith-Lemli-Opitz syndrome, the precursor 7DHC is potentially toxic in high concentrations, and cholesterol deficiency is almost certainly detrimental.
Smith, Lemli, and Opitz initially described Smith-Lemli-Opitz syndrome (while working at the University of Wisconsin) as a genetic MCA/ID syndrome, in 1964. [4] They named the condition RSH after the first initial of the last names of the first 3 patients ascertained. [5] The clinical characteristics of Smith-Lemli-Opitz syndrome have been well established over the past 4 decades.
The etiology of Smith-Lemli-Opitz syndrome was unknown until 1993, when Irons et al discovered that patients with Smith-Lemli-Opitz syndrome had low plasma cholesterol levels and accumulated sterol precursors such as 7DHC. [6] A deficiency of the microsomal enzyme DHCR7, which reduces the 7-8 double bond of 7DHC to form cholesterol in the final step of the cholesterol synthetic pathway, was hypothesized and later proven to cause Smith-Lemli-Opitz syndrome. Mutations in the DHCR7 gene are responsible for Smith-Lemli-Opitz syndrome. Therefore, Smith-Lemli-Opitz syndrome can now be considered a classic inborn error of metabolism.
Currently, the reason defects in cholesterol synthesis cause congenital malformations is not known. Several disparate lines of research have led to recent understanding of the critical and somewhat unexpected role of cholesterol in early human development. Cholesterol is important in cell membranes, serves as the precursor for steroid hormones and bile acids, and is a major component in myelin. Cholesterol is covalently bound to the embryonic signaling protein sonic hedgehog (Shh) in a necessary step of the autoprocessing of the precursor to active form, occurring about gestational day 0-7 in humans.
Shh plays a critical role in several embryologic fields relevant to Smith-Lemli-Opitz syndrome (eg, brain, face, heart, limbs). Therefore, cholesterol is an essential triggering agent in the early developmental program of the human. Because 7DHC can also activate Shh, cholesterol deficiency that leads to decreased activation of Shh is probably not the sole explanation for congenital malformations in this syndrome.
Abnormalities in the Shh-patched signaling cascade presumably play a role. Membrane instability and dysmyelination from cholesterol deficiency and accumulation of 7DHC and other potentially toxic cholesterol precursors are also likely to contribute to the Smith-Lemli-Opitz syndrome phenotype.
Increased isoprenoids were reported in Smith-Lemli-Opitz syndrome, but the role these nonsterol isoprenoids play in the pathophysiology of this disorder is unclear. [7]
A study by Merkens et al indicated that high 7DHC plasma levels correlate with feeding difficulties in patients with Smith-Lemli-Opitz syndrome. In the report, which involved 26 patients (aged 0.4-19 years) with the syndrome, the investigators found that patients with a plasma level of more than 0.24 mmol/L or a cholesterol concentration of less than 1.95 mmol/L were more likely to require use of a gastrostomy tube. [8]
A study by Sparks et al found reduced levels of the neurotransmitter metabolites 5-hydroxyindoleacetic acid (from serotonin) and homovanillic acid (from dopamine) in the cerebrospinal fluid of patients with Smith-Lemli-Opitz syndrome. The investigators suggested that this may result from a sterol-associated defect in synaptic vesicle development. [9]
Epidemiology
Frequency
United States
Prevalence of Smith-Lemli-Opitz syndrome has been estimated to be 1 in 20,000-60,000 births among Caucasians. Smith-Lemli-Opitz syndrome is also not uncommon in Hispanics. Its specific prevalence in different populations has not been precisely determined. The higher-than-expected prevalence of Smith-Lemli-Opitz syndrome suggests a heterozygote advantage.
Only one description of an African-American patient has been published, although no biochemical or molecular confirmation of Smith-Lemli-Opitz syndrome was available. [10] In a study of 150 biochemically diagnosed patients with Smith-Lemli-Opitz syndrome, only one individual was of African descent. [11] In 2000, Yu and colleagues did not detect the mutation among 121 Africans from Sierra Leone. [12] In 2001, Nowaczyk and colleagues reported an IVS8-1G>C (common Smith-Lemli-Opitz syndrome mutation) carrier frequency of 1.09% (17 per 1559 population) in Canadian whites and 0.79% (4 per 504 population) in Canadians of African descent; however, no African Canadian patients were identified. [13]
The results of Wright et al's 2003 study indicate an IVS8-1G>C carrier frequency of 0.73% (10 per 1378 population) in African Americans. [14] This predicts the prevalence of Smith-Lemli-Opitz syndrome due to IVS8-1G>C homozygosity to be 1 case per 75,061 persons in the African American population. Although the African American carrier frequency of the IVS8-1G>C allele was determined to be 0.73%, few African American patients with Smith-Lemli-Opitz syndrome have been identified.
Carrier frequency for Smith-Lemli-Opitz syndrome is approximately 1 in 30 persons of northern European descent, suggesting a disease frequency of 1 per 5000-18,000 people. The actual disease prevalence may be lower because of fetal losses and missed diagnoses or misdiagnoses at the most severe and most mild ends of the severity spectrum.
International
Smith-Lemli-Opitz syndrome has been described in patients from the United States, many northern European countries, Japan, South America, and other countries. Smith-Lemli-Opitz syndrome appears to be uncommon in Japan. The frequency of Smith-Lemli-Opitz syndrome appears to be similar in northern Europe and the United States, but additional studies are needed to determine the frequency of Smith-Lemli-Opitz syndrome in other regions. The European origin of some of the major mutations found in Smith-Lemli-Opitz syndrome has been explored. [15]
Mortality/Morbidity
Spontaneous abortion of fetuses with Smith-Lemli-Opitz syndrome is not unusual. Stillbirths have also been reported, although a prospective study by Gibbins et al found that homozygous DHCR7 mutations (indicative of Smith-Lemli-Opitz syndrome) were present in only one out of 139 unexplained stillbirths (0.7%), suggesting that unrecognized DHCR7 mutations are not strongly associated with stillbirth. [16]
A study by Daum et al indicated that homozygosity for the founder variant DHCR7:c.964-1G>C, carried in the Ashkenazi Jewish population and other populations, is associated with severe phenotype for Smith-Lemli-Opitz syndrome, including early miscarriage in some cases, and a very high rate of early postnatal mortality. [17]
Death from multiorgan system failure during the first weeks of life is typical in the most severely affected individuals with Smith-Lemli-Opitz syndrome. Cause of death can include pneumonia, lethal congenital heart defect, or hepatic failure. Survival is unlikely if the plasma cholesterol level is less than approximately 20 mg/dL as measured by gas chromatography, which is used because routine methods of cholesterol measurement include precursor sterols.
Congenital heart disease is not uncommon in Smith-Lemli-Opitz syndrome and can cause cyanosis or congestive heart failure. Pulmonary hypertension has been noted in at least one patient. [18] Vomiting, feeding difficulties, constipation, toxic megacolon, electrolyte disturbances, and failure to thrive are common and, in some cases, related to GI anomalies. Liver disease has been described in a subset of patients. [19] Visual loss may occur because of cataracts, optic nerve abnormalities, or other ophthalmologic problems, [20] and subclinical retinal abnormalities may be noted on electroretinography. [21] Cataracts may occur acutely in the postnatal period. [22] Hearing loss is fairly common.
Race
See Frequency.
Sex
As an autosomal recessive genetic condition, Smith-Lemli-Opitz syndrome is equally prevalent among males and females.
Age
Smith-Lemli-Opitz syndrome is a genetic condition that is present from conception, but signs may occasionally be so subtle that patients avoid detection until later childhood or even adulthood. Some have postulated that the mildest cases may completely escape detection in some instances. More commonly, Smith-Lemli-Opitz syndrome is suspected at birth or shortly thereafter because of birth defects.
-
Child with Smith-Lemli-Opitz syndrome.





