Practice Essentials
Potter syndrome refers to the typical physical appearance and associated pulmonary hypoplasia of a neonate as a direct result of oligohydramnios and compression while in utero. See the image below. The renal function and respiratory status of neonates born with Potter syndrome must be assessed. Once the long-term prognosis of survival is determined, resuscitation and management plans should be addressed.
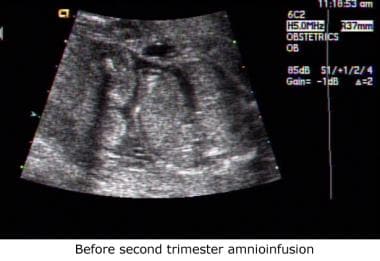 Sonogram obtained before second-trimester amnioinfusion. This fetus has bilaterally absent kidneys consistent with a diagnosis of Potter syndrome. The cystic structures in the renal fossae are most likely the adrenal glands.
Sonogram obtained before second-trimester amnioinfusion. This fetus has bilaterally absent kidneys consistent with a diagnosis of Potter syndrome. The cystic structures in the renal fossae are most likely the adrenal glands.
Signs and symptoms
Findings at physical examination may include the following:
-
Potter facies (flattened nose, recessed chin, prominent epicanthal folds, and low-set abnormal ears)
-
Pulmonary hypoplasia
-
Features of Eagle-Barrett (prune belly) syndrome (deficient abdominal wall, undescended testes, dilated ureters, and a renal pelvis)
-
Skeletal malformations (hemivertebrae, sacral agenesis, and limb anomalies)
-
Ophthalmologic malformations (cataract, angiomatous malformation in the optic disc area, prolapse of the lens, and expulsive hemorrhage)
-
Cardiovascular malformations (ventricular septal defect, endocardial cushion defect, tetralogy of Fallot, and patent ductus arteriosus)
See Presentation for more detail.
Diagnosis
Laboratory studies
The following tests are recommended in patients with suspected Potter syndrome:
-
Measurement of serum electrolyte levels
-
Measurement of serum creatinine levels
-
Complete blood cell (CBC) count with differential
-
Urinalysis
If sepsis is suspected, obtain cultures of the urine, blood, and cerebrospinal fluid. Chromosomal analysis is obtained if the physical examination findings suggest an associated genetic disorder.
Imaging studies
Prenatal imaging studies may include the following:
-
Abdominal and transvaginal ultrasonography
-
Doppler ultrasonography
-
Antenatal MRI
-
Amnioinfusion
Neonatal imaging studies may include the following:
-
Abdominal ultrasonography
-
Chest radiography
-
Voiding cystourethrography
-
Nuclear renal scanning
See Workup for more detail.
Management
Multiple medications are typically indicated in patients with acute or chronic renal failure. The treatment should address fluid and electrolyte disturbances, hypertension, anemia, calcium and phosphorus disorders, and growth failure.
See Treatment for more detail.
Background
The term Potter syndrome was coined after the pathologist Edith Potter, who in 1946 described the facial characteristics of infants with bilateral renal agenesis. [1] From her research, she was able to deduce the sequence of events that lead to these features. Other conditions resulting in oligohydramnios, such as obstructive uropathy, cystic kidney diseases, renal hypoplasia, and premature rupture of membranes lead to the same clinical findings. Hence, the terms Potter sequence or oligohydramnios sequence emerged. Regardless of the root cause for oligohydramnios, the terms Potter syndrome, Potter sequence, and oligohydramnios sequence are used interchangeably in the published literature.
Pathophysiology
Prior to 16 weeks' gestation, the amount of amniotic fluid is dependent on the transmembrane flow. After that, fetal urine production is the predominant mechanism that determines the amniotic fluid volume. The fetus continuously swallows amniotic fluid, which is reabsorbed by the GI tract and then reintroduced into the amniotic cavity by the kidneys. Oligohydramnios occurs if the volume of amniotic fluid is less than normal for the corresponding period of gestation. This may be due to decreased urine production secondary to bilateral renal agenesis, obstruction of the urinary tract, or, occasionally, prolonged rupture of membranes. [2, 3] The resulting oligohydramnios is the cause of the deformities observed in Potter syndrome. The mechanism of lung hypoplasia in this condition is not clear. It is believed that adequate space in the fetal thorax and the movement of amniotic fluid into the fetal lungs are required for the normal development of lungs.
Genetics
The genetic aspect of renal malformation has not been fully understood yet, and there is much current research work in this field.
During nephrogenesis, multiple genes, transcription factors, and growth factors control the essential interaction between the ureteric bud and the metanephric mesenchyme. For example, the Lim1 and Pax2 transcription factors are essential for the formation of the mesonephric duct, from which the ureteric bud develops. Lim1 -deficient mice have complete renal agenesis. If the Pax2 gene is deficient, there is deletion of the caudal portion of the mesonephric duct, which results in renal agenesis. [4, 5]
WT-1, a zinc-finger transcription factor expressed in the metanephric mesenchyme, is essential for ureteric bud outgrowth. Homozygous null-mutants for WT-1 have complete renal agenesis. Similarly, transcription factor EYA1 from the metanephric mesenchyme is required for ureteric bud outgrowth, and deficiency of this protein is shown to cause branchio-oto-renal syndrome. [6]
The glial cell line–derived neurotrophic factor (GDNF) from the metanephric mesenchyme binds to the C-ret receptor on the branching ureteric bud and is responsible for the branching and elongation of the ureteric bud. Inactivation of either GDNF or the C-ret receptor leads to renal agenesis. Heterozygotes may have a unilateral renal abnormality, whereas the contralateral kidney has normal development. [7]
Limb deformity (ld) gene codes for 4 different spliced formin genes, which are expressed in the mesonephric duct and branching ureteric ducts. Mutation of the ld gene leads to limb deformity with renal agenesis. Mutation of the formin IV gene leads only to kidney abnormalities. Homozygous mutation of the alpha-8 integrin subunit produces abnormalities similar to ld mutation with deformities including renal aplasia, dysplasia, or hypoplasia. [8]
Transcription factors such as EMX-2, BF-2, fibroblast growth factor 7 (FGF 7), epithelial growth factor receptor (EGF-R), GDNF, retinoic acid receptor alpha, and beta 2 are involved in the branching of the ureteric bud. A heterozygous mutation defect of the growth factor bone morphogenetic protein 4 (bmp 4) leads to renal hypoplasia or dysplasia, ureterovesicular junction obstruction, hydronephrosis, or the bifid/duplex kidney. This is a defect of ureteric branching and not induction of the ureteric bud; thus, renal aplasia does not occur. [9, 10, 11]
The autosomal recessive mutations of genes in the renin-angiotensin pathway result in renal tubular dysgenesis due to failed development of proximal tubules. These are heterogeneous mutations of renin, angiotensin, angiotensin converting enzyme, or type 1 angiotensin II receptor. [12, 13] Some reports have suggested that the clinical picture of renal tubular dysgenesis is similar to the infants born to mothers who had received angiotensin-converting enzyme inhibitors or angiotensin II receptor blockers during pregnancy. [14, 15]
Hepatocyte nuclear factor (HNF)-1beta gene (TCF2) is normally expressed in the Wolffian duct, metanephric tubules, and Mullerian during fetal life. This gene, which was originally described in relation to maturity-onset diabetes, has been now recognized as a cause of cystic renal dysplasia. [16, 17, 4] Uroplakins IIIa is a protein expressed on the mammalian urothelia and has been suggested to be involved in defects of early kidney development such as renal hypoplasia/dysplasia. [18]
Recognized genetic disorders such as renal coloboma syndrome (PAX2 mutation) and branchio-oto-renal syndrome (EYA1 mutation) are therefore associated with renal agenesis or dysplastic kidney abnormalities. [6, 19]
Some of the other reported mutations associated with renal hypodysplasia are FREM 1 (which causes bifid nose, renal agenesis, and anorectal malformation), [20] FRAS 1/FREM 2 (Fraser syndrome), [21, 22, 23] LRP4 (Cenani-Lenz syndrome), [24] GLI3 (Pallister-Hall syndrome), [25] SALL1 (Townes-Brocks syndrome, which has triad of imperforate anus, dysplastic ears, and thumb malformation), [26] KAL1 (Kallman syndrome), [27] and GATA3 (renal hypodysplasia, which is associated with hypothyroidism and sensory-neural deafness). [28]
Cases have been reported of families with both unilateral and bilateral renal agenesis. This condition, termed hereditary renal adysplasia (HRA), is an autosomal dominant trait with incomplete penetrance and variable expression. Associated non-urogenital anomalies have been reported in HRA. [29]
Etiology
Causes of Potter syndrome may include the following:
-
Bilateral renal agenesis
-
Cystic kidney diseases
Autosomal recessive polycystic kidney disease
Autosomal dominant polycystic kidney disease
Multicystic renal dysplasia
-
Obstructive uropathy
-
Early rupture of membranes
No preventive measures are known for any causes listed above.
A retrospective analysis of children with Potter syndrome found that 21% had bilateral renal agenesis, 47% had cystic dysplasia, 25% had obstructive uropathy, and 5% had other defects. Posterior urethral valves were the most common cause of bladder outlet obstruction (60%). [30]
Epidemiology
United States data
Potter syndrome is mostly associated with obstruction of the urinary tract or severe bilateral renal hypoplasia. Bilateral renal agenesis is estimated to occur in about 1 of 5000 fetuses [31] and is responsible for 20% of Potter syndrome cases. The frequency of other causes of Potter syndrome is not known. The associated maternal high-risk factors for bilateral renal agenesis are maternal body mass index greater than 30, smoking, and binge drinking. [32, 33]
International data
Data from 20 registries of 12 European countries was collected on 709,030 livebirths, stillbirths, and induced abortions. Bilateral renal agenesis was seen in 95 cases and out of this prenatal diagnosis was made in 86 cases. [34] In one other study from Europe's 17 registries reported 4366 cases diagnosed with 11 severe congenital malformations, out of which 257 cases had bilateral renal agenesis. [35]
Race-, sex-, and age-related demographics
No racial predilection is known. Males have an increased incidence of the Potter syndrome because they have a higher rate of Eagle-Barrett (prune belly) syndrome [36] and obstructive uropathy secondary to posterior urethral valves. Patients present as neonates.
Prognosis
Morbidity/mortality
Potter syndrome is usually fatal in the first few days of the patient's life; most often, the cause is pulmonary failure. Bilateral renal agenesis is incompatible with extrauterine life, and 33% of fetuses die in utero. A 70% survival rate has been reported among 23 infants with antenatal oligohydramnios and pulmonary hypoplasia. [37] The primary disease in these 23 infants included obstructive uropathy, autosomal recessive polycystic kidney disease, renal tubular dysgenesis, and bilateral renal dysplasia.
Neonates with the milder form of Potter syndrome have an increased morbidity rate because of respiratory failure, pneumothorax, and acute renal failure during the neonatal period. During early childhood, patients may have chronic lung disease and chronic renal failure.
A number of abnormalities are associated with bilateral renal agenesis, such as caudal dysgenesis, VATERL (V ertebral anomalies, A nal atresia, C ardiac defects, T racheoesophageal fistula, R enal defects, L imb defects), [38] caudal dysplasia syndrome, and isolated anomalies of the cardiovascular, skeletal, and central nervous systems. [39, 40, 41, 42] These abnormalities can add to the morbidity and increased mortality in these patients.
Complications
Associated pulmonary complications include the following:
-
Spontaneous pneumothorax due to pulmonary hypoplasia
-
Neonatal respiratory distress due to pulmonary hypoplasia
Associated renal complications include the following:
-
Hypertension that requires antihypertensive drug therapy
-
Hyperkalemia
-
Hypocalcemia
-
Hyperphosphatemia
-
Hyponatremia
-
Acute renal failure
Patient Education
Prenatal care should be provided with the help of a perinatologist. Parents should be fully aware of and educated about oligohydramnios and its long-term consequences on the developing fetus.
-
Sonogram obtained before second-trimester amnioinfusion. This fetus has bilaterally absent kidneys consistent with a diagnosis of Potter syndrome. The cystic structures in the renal fossae are most likely the adrenal glands.





