Practice Essentials
Neuroblastoma is the most common extracranial solid tumor of infancy. It is an embryonal malignancy of the sympathetic nervous system arising from neuroblasts (pluripotent sympathetic cells). In the developing embryo, these cells invaginate, migrate along the neuraxis, and populate the sympathetic ganglia, adrenal medulla, and other sites. The patterns of distribution of these cells correlate with the sites of primary neuroblastoma presentation.
Histologic subtypes of neuroblastoma are shown in the image below.
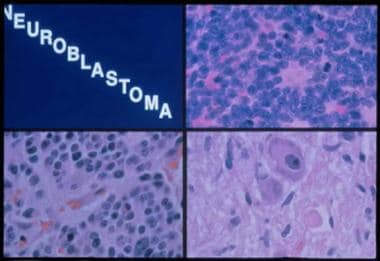 Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
Signs and symptoms
Signs and symptoms of neuroblastoma vary with the site of presentation. Generally, symptoms include the following:
-
Abdominal pain
-
Emesis
-
Weight loss
-
Anorexia
-
Fatigue
-
Bone pain
See Presentation for more detail.
Diagnosis
Laboratory studies
Recommended laboratory studies include the following:
-
Complete blood cell (CBC) count and differential
-
Urine collection for catecholamines and urinalysis
-
Serum creatinine
-
Liver function tests
-
Prothrombin time/activated prothrombin time
-
Electrolytes
-
Calcium, magnesium, phosphorus
-
Uric acid
-
Serum lactate dehydrogenase
-
Ferritin
-
Thyroid-stimulating hormone, T4
-
Immunoglobulin (Ig)G levels
Imaging studies
The following studies may be indicated in patients with neuroblastomas:
-
Chest and abdominal radiographs
-
A computed tomography (CT) scan of the primary site
-
Magnetic resonance imaging (MRI) in patients with paraspinal masses or Horner syndrome
-
I 123/ 131-methyliodobenzylguanadine (MIBG) scanning
-
A technetium-99 bone scan
-
Skeletal surveys
See Workup for more detail.
Management
Age, stage, and biological features encountered in tumor cells are important prognostic factors and are used for risk stratification and treatment assignment. The differences in outcome for patients with neuroblastoma are striking. Patients with low-risk and intermediate-risk neuroblastoma have excellent prognosis and outcome. However, those with high-risk disease continue to have very poor outcomes despite intensive therapy. Unfortunately, approximately 70-80% of patients older than 18 months present with metastatic disease, usually in the lymph nodes, liver, bone, and bone marrow. Less than half of these patients are cured, even with the use of high-dose therapy followed by autologous bone marrow or stem cell rescue.
See Treatment and Medication for more detail.
Pathophysiology
Chromosomal and molecular markers
Over the last 2 decades, many chromosomal and molecular abnormalities have been identified in patients with neuroblastoma. These biologic markers have been evaluated to determine their value in assigning prognosis, and some of these have been incorporated into the strategies used for risk assignment.
The most important of these biologic markers is MYCN. MYCN is an oncogene that is overexpressed in approximately one quarter of cases of neuroblastoma via the amplification of the distal arm of chromosome 2. This gene is amplified in approximately 25% of de novo cases and is more common in patients with advanced-stage disease. Patients whose tumors have MYCN amplification tend to have rapid tumor progression and poor prognosis, even in the setting of other favorable factors such as low-stage disease or 4S disease.
In contrast to MYCN, expression of the H-ras oncogene correlates with lower stages of the disease. Cytogenetically, the presence of double-minute chromatin bodies and homogeneously staining regions correlates with MYCN gene amplification. Deletion of the short arm of chromosome 1 is the most common chromosomal abnormality present in neuroblastoma and confers a poor prognosis. The 1p chromosome region likely harbors tumor suppressor genes or genes that control neuroblast differentiation. Deletion of 1p is more common in near-diploid tumors and is associated with a more advanced stage of the disease. Most of the deletions of 1p are located in the 1p36 area of the chromosome.
A relationship between 1p loss of heterozygosity (LOH) and MYCN amplification has been described. Other allelic losses of chromosomes 11q, 14q, and 17q have been reported, suggesting that other tumor suppressor genes may be located in these chromosomes. Loss of heterozygosity at 11q23 has been described and is an independent prognostic factor. Another characteristic of neuroblastoma is the frequent gain of chromosome 1.
DNA index is another useful test that correlates with response to therapy in infants. Look et al. demonstrated that infants whose neuroblastoma have hyperdiploidy (ie, DNA index >1) have a good therapeutic response to cyclophosphamide and doxorubicin. [1] In contrast, infants whose tumors have a DNA index of 1 are less responsive to the latter combination and require more aggressive therapy. DNA index does not have any prognostic significance in older children. In fact, hyperdiploidy in children more frequently occurs in the context of other chromosomal and molecular abnormalities that confer a poor prognosis.
Three neurotrophin receptor gene products, TrkA, TrkB, and TrkC, are tyrosine kinases that code for a receptor of members of the nerve growth factor (NGF) family. Their ligands include p75 neurotrophin receptor (p75NTR) NGF, and brain-derived neurotrophic factors (BDNFs). Interestingly, TrkA expression is inversely correlated with the amplification of the MYCN gene, and the expression of the TrkC gene is correlated with TrkA expression. In most patients younger than 1 year, a high expression of TrkA correlates with a good prognosis, especially in patients with stages 1, 2, and 4S. In contrast, TrkB is more commonly expressed in tumors with MYCN amplification. This association may represent an autocrine survival pathway.
Disruption of normal apoptotic pathways may also play a role in neuroblastoma pathology. Disruption of these normal pathways may play a role in therapy response as a result of epigenetic silencing of gene promoters in apoptotic pathways. Drugs that target DNA methylation, such as decitabine, are being explored in preliminary studies.
Other biologic markers associated with poor prognosis include increased levels of telomerase RNA and lack of expression of glycoprotein CD44 on the tumor cell surface. P-glycoprotein (P-gp) and multidrug resistance protein (MRP) are 2 proteins expressed in neuroblastoma. These proteins confer a multidrug-resistant (MDR) phenotype in some cancers. Their role in neuroblastoma is controversial. Reversal of MDR is one target for novel drug development.
A study by Challagundla et al reported that exosomic microRNAs released within the neuroblastoma environment affect resistance to chemotherapy. [2]
Anatomic
Origin and migration pattern of neuroblasts during fetal development explains the multiple anatomic sites where these tumors occur; location of tumors varies with age. Tumors can develop in the abdominal cavity (40% adrenal, 25% paraspinal ganglia) or other sites (15% thoracic, 5% pelvic, 3% cervical tumors, 12% miscellaneous). Infants more commonly present with thoracic and cervical tumors, whereas older children more frequently have abdominal tumors.
Most patients present with signs and symptoms related to tumor growth, although small tumors have been detected due to the common use of prenatal ultrasonography. Large abdominal tumors often result in increased abdominal girth and other local symptoms (eg, pain). Paraspinal dumbbell tumors can extend into the spinal canal, impinge on the spinal cord, and cause neurologic dysfunction.
Stage of the tumor at the time of diagnosis and age of the patient are the most important prognostic factors. Although patients with localized tumors (regardless of age) have an excellent outcome (80-90% 3-year event-free survival [EFS] rate), patients older than 18 months with metastatic disease fare poorly. Generally, more than 50% of patients present with metastatic disease at the time of diagnosis, 20-25% have localized disease, 15% have regional extension, and approximately 7% present during infancy with disseminated disease limited to the skin, liver, and bone marrow (stage 4S).
Physiologic and biochemical
More than 90% of patients have elevated homovanillic acid (HVA) and/or vanillylmandelic acid (VMA) levels detectable in urine. Mass screening studies using urinary catecholamines in neonates and infants in Japan, Quebec, and Europe have demonstrated the ability to detect neuroblastoma before it is clinically apparent. However, most of the tumors identified occur in infants with a good prognosis. None of these studies shows that mass screening decreases deaths due to high-risk neuroblastoma.
Markers associated with a poor prognosis include (1) elevated ferritin levels, (2) elevated serum lactate dehydrogenase (LDH) levels, and (3) elevated serum neuron-specific enolase (NSE) levels. However, these markers have become less important due to the discovery of more relevant biomarkers (ie, chromosomal and molecular markers). In fact, ferritin was not included in the recent formulation of the International Neuroblastoma Risk Group Classification System because it was not found to be of prognostic difference in the high-risk group.
Histologic
Pluripotent sympathetic stem cells migrate and differentiate to form the different organs of the sympathetic nervous system. The normal adrenal gland consists of chromaffin cells, which produce and secrete catecholamines and neuropeptides. Other cells include sustentacular cells, which are similar to Schwann cells, and scattered ganglion cells. Histologically, neural crest tumors can be classified as neuroblastoma, ganglioneuroblastoma, and ganglioneuroma, depending on the degree of maturation and differentiation of the tumor.
The undifferentiated neuroblastomas histologically present as small, round, blue cell tumors with dense nests of cells in a fibrovascular matrix and Homer-Wright pseudorosettes. These pseudorosettes, which are observed in 15-50% of tumor samples, can be described as neuroblasts surrounding eosinophilic neuritic processes. The typical tumor shows small uniform cells with scant cytoplasm and hyperchromatic nuclei. A neuritic process, also called neuropil, is a pathognomonic feature of neuroblastoma cells. NSE, chromogranin, synaptophysin, and S-100 immunohistochemical stains are usually positive. Electron microscopy can be useful because ultrastructural features (eg, neurofilaments, neurotubules, synaptic vessels, dense core granules) are diagnostic for neuroblastoma.
In contrast, the completely benign ganglioneuroma is typically composed of mature ganglion cells, Schwann cells, and neuritic processes, whereas ganglioneuroblastomas include the whole spectrum of differentiation between pure ganglioneuromas and neuroblastomas. Because of the presence of different histologic components, the pathologist must thoroughly evaluate the tumor; the regions with different gross appearance may demonstrate a different histology.
Neuroblastic nodules are present in the fetal adrenal gland and peak at 17-18 weeks' gestation. Most of these nodules spontaneously regress and likely represent remnants of fetal development. Some of these may persist and lead to the development of neuroblastoma.
Shimada histopathologic classification system
Shimada et al developed a histopathologic classification in patients with neuroblastoma. [3] This classification system was retrospectively evaluated and correlated with outcome in 295 patients with neuroblastoma who were treated by the Children's Cancer Group (CCG). Important features of the classification include (1) the degree of neuroblast differentiation, (2) the presence or absence of Schwannian stromal development (stroma-rich, stroma-poor), (3) the index of cellular proliferation (known as mitosis-karyorrhexis index [MKI]), (4) nodular pattern, and (5) age. Using these components, patients can be classified into the following histology groups:
Favorable histology group includes the following:
-
Patients of any age with stroma-rich tumors without a nodular pattern
-
Patients younger than 18 months with stroma-poor tumors, an MKI of less than 200/5000 (200 karyorrhectic cells per 5000 cells scanned), and differentiated or undifferentiated neuroblasts
-
Patients younger than 60 months with stroma-poor tumors, an MKI of less than 100/5000, and well-differentiated tumor cells
Unfavorable histology group includes the following:
-
Patients of any age with stroma-rich tumors and a nodular pattern
-
Patients of any age with stroma-poor tumors, undifferentiated or differentiated neuroblasts, and an MKI more than 200/5000
-
Patients older than 18 months with stroma-poor tumors, undifferentiated neuroblasts, and an MKI more than 100/5000
-
Patients older than 18 months with stroma-poor tumors, differentiated neuroblasts, and an MKI of 100-200/5000
-
Patients older than 60 months stroma-poor, differentiated neuroblasts, and an MKI less than 100
Shimada et al’s original classification was adopted and integrated into the International Neuroblastoma Pathology Classification (INPC). This was most recently revised. [4] The INPC system remains age-dependent.
Etiology
The cause of neuroblastoma is unknown, and no specific environmental exposure or risk factors have been identified.
Because of young age of onset with this disease, investigators have focused on events before conception and during gestation.
According to SEER data, factors investigated for which evidence is limited or inconsistent include medications, hormones, birth characteristics, congenital anomalies, previous spontaneous abortion or fetal death, alcohol or tobacco use, and paternal occupational exposures.
The vast majority of neuroblastoma arises sporadically without family history of the disease. However, 1-2% of newly diagnosed cases do have a family history of neuroblastoma. Patients with familial neuroblastoma often present at earlier age or with several distinct primary tumors.
Neuroblastoma has been known to occur in the setting of other disorders that are linked to abnormal development of neural crest tissues, such as Hirschsprung disease or central congenital hypoventilation syndrome. Genome-wide analysis of neuroblastoma from these rare familial cases has identified a genetic defect involved in these cases. Cases of neuroblastoma that accompany other congenital abnormalities of the neural crest have been associated with a germline mutation in PHOX2B. This gene is a homeobox gene that acts as a regulator of autonomic nervous system development.
In familial neuroblastoma cases that are not associated with other congenital disorders of neural crest development, ALK mutations have been identified in the germline. [5] These mutations largely occur in the kinase domain causing activation of ALK signaling. Efforts are ongoing to investigate the incidence of ALK mutations across all subsets of neuroblastoma, but initial evidence indicates that somatic mutations of the ALK gene are also present in some cases of sporadic neuroblastoma.
A 2014 study reported that deep-sequencing techniques can identify new ALK mutations at relapse of neuroblastoma, suggesting that patients would benefit from repeated tumor sampling. [6]
De Brouwer et al illustrate the occurrence of the ALK mutation specifically in neuroblastomas. Although they studied a small proportion of cases, mutations were found in similar frequencies in favorable and unfavorable outcome cases. The F1174L mutant was found more frequently in the poor outcome subgroup. [7] This example illustrates the heterogeneity of cancer and the likely possibility that targeted therapies to the ALK gene may be of benefit in a subset of ALK cancers, which may possibly include a small subset of MYC-amplified neuroblastomas. The challenge for drug development in neuroblastoma is to identify upfront high-risk cases that may benefit from ALK-directed therapy.
Genome-wide association studies (GWAS) have been used to discover numerous genetic variations associated with neuroblastoma. Variations in LMO1, BARD1, and FLJ22536 have all been associated with aggressive forms of neuroblastoma. [8, 9, 10] Genomic variations within DUSP12, DDX4, IL21RA, and HSD17B12 are associated with low-risk forms of neuroblastoma. [11]
Epidemiology
United States statistics
Neuroblastoma accounts for approximately 6% of childhood cancers in the United States. [12] About 650 new cases are diagnosed in the United States each year. According to the Surveillance, Epidemiology, and End Report (SEER), incidence is approximately 9.5 cases per million children. [13]
International statistics
The incidence in other industrialized nations appears to be similar to that observed in the United States. International reports have shown that the incidence rates of neuroblastoma are highest among high income countries in Europe and North America, and lower in low income countries in Africa, Asia, and Latin America. No published data are available on the incidence in the emerging high-income countries of Asia. [14]
Race-, sex-, and age-related demographics
The incidence of neuroblastoma is higher in White children than in Black children. However, race does not appear to have any effect on outcome.
Males have a slightly higher incidence of neuroblastoma than females, with a male-to-female ratio of 1.2:1.
Age distribution is as follows: 40% of patients are younger than 1 year when diagnosed, 35% are aged 1-2 years, and 25% are older than 2 years when diagnosed. According to SEER, incidence decreases every consecutive year up to age 10 years, after which the disease is rare. [13]
Prognosis
Determinants of response and outcome include the following:
-
Stage, age, and several biologic characteristics of the tumor determine outcome.
-
Similarly, the patient may also have genetic polymorphism characteristics that influence drug absorption, distribution, metabolism, and excretion.
The following treatment strategies are available to treat patients with recurrent neuroblastoma:
-
A local recurrence in a patient with low-stage disease generally has a good prognosis, and patients usually receive standard chemotherapy, surgery, and/or radiation as necessary.
-
Patients with disseminated disease at presentation have a high recurrence rate and a poor outcome.
-
For patients with recurrent disease in this setting, various phase I/II agents are generally available.
The following response criteria are used to evaluate the efficacy of therapy:
-
Complete clinical response - More than 90% decrease (sum of the products of the greatest perpendicular diameters) of the primary tumor and metastatic disease (if any), no new lesions, healing of bone lesions
-
Partial clinical response - A decrease of 50% or less (sum of the products of the greatest perpendicular diameters) of the primary tumor and metastatic disease (if any), no new lesions, healing of bone lesions
-
Minor response - More than 25% and less than 50% decrease (sum of the products of the greatest perpendicular diameters) of primary tumor and metastatic disease (if any), no new lesions, healing of bone lesions
-
No response - Less than 25% decrease (sum of the products of the greatest perpendicular diameters) of primary tumor or metastatic disease (if any), no new lesions
-
Progressive disease - More than 25% increase (sum of the products of the greatest perpendicular diameters) of the primary tumor or all metastatic lesions (if any), appearance of new lesions
Morbidity/mortality
According to the SEER data, the overall 5-year survival rate for children with neuroblastoma has improved from 24% in 1960-1963 to 55% in 1985-1994. [13] In part, this increase in survival rate may be due to better detection of low-risk tumors in infants. The survival rate 5 years from diagnosis is approximately 83% for infants, 55% for children aged 1-5 years, and 40% for children older than 5 years. Improvements in diagnostic imaging modalities, medical and surgical management, and supportive care have contributed to the improved survival rates. [15]
Most patients with neuroblastoma present with disseminated disease, which confers a poor prognosis and is associated with a high mortality rate. Tumors in these patients usually have unfavorable pathologic and/or molecular features. The 3-year EFS for high-risk patients treated with conventional chemotherapy, radiation therapy, and surgery is less than 20%. Differentiating agents and dose intensification of active drugs, followed by autologous bone marrow transplant, have been reported to improve the outcome for these patients, contributing to an EFS of 38%. A recent single-arm study of tandem stem cell transplantation reported a 3-year EFS of 58%, but randomized studies of this approach are ongoing. [16]
Morbidity of high-dose chemotherapy approaches can be substantial, although the treatment-related mortality rates have decreased with improvements in supportive care and hematopoietic support with growth factors and stem cells instead of bone marrow.
Complications
The following complications may occur:
-
The most worrisome complication at disease presentation is cord compression from a paraspinal tumor. Evaluation of the patient by a neurosurgeon and consultation with a radiation oncologist are important.
-
In some individuals with neuroblastomas, early institution of chemotherapy is accepted if the tumor can be biopsied within 72 hours to make a diagnosis and to obtain necessary biologic studies. In the acute setting, chemotherapy may be as efficient as radiotherapy or laminectomy, and it may cause less morbidity. Treatment of cord compression with chemotherapy and steroids usually results in less complications; however, radiation therapy or surgery is often used as front-line treatment to prevent impending or progressive neurologic damage. In children who present with significant neurologic symptoms, none of these interventions assure a return of normal neurologic (motor) function.
-
Tumor lysis syndrome is unusual in neuroblastoma
-
Patients may present with severe hypertension or renal insufficiency, making initiation of chemotherapy, especially with platinum drugs, more difficult.
-
Myelosuppression and immunosuppression place the patient at risk of bleeding and infection. Febrile neutropenia is a medical emergency and requires immediate admission to the hospital and initiation of broad-spectrum antibiotic treatment.
-
After several cycles of therapy, depending on drugs administered, patients may develop impaired renal function, hearing loss, or delayed count recovery.
Patient Education
For compliance and good medical care, patients and families must understand the importance of treatment and adverse effects of medications used. In addition, they should learn to recognize and identify signs and symptoms of complications that require urgent medical care.
For further information for patients and their families, see the WebMD Medical Reference article What Is Neuroblastoma?
-
Histologic subtypes of neuroblastoma. Top right panel, neuroblastoma: A monotonous population of hyperchromatic cells with scant cytoplasm. Bottom left panel, ganglioneuroblastoma: Increased schwannian stroma. Bottom right panel, ganglioneuroma: Mature ganglion cell with schwannian stroma.
-
CT scan of abdomen in a patient with a retroperitoneal mass arising from the upper pole of the left kidney and elevated urine catecholamines.
-
MRI of a left adrenal mass. The mass was revealed by fetal ultrasonography at 30 weeks' gestation. During infancy, the mass was found on the inferior pole of the left adrenal and was completely resected. Before surgery, the metastatic workup was negative. Surgical pathology service confirmed a diagnosis of neuroblastoma. After 3 years of follow-up care, no recurrence was observed.
-
A one-week-old neonate had abdominal ultrasonography for evaluation of projectile vomiting. A right adrenal mass (100% cystic) was an incidental finding. Evaluation of the mass by CT was consistent with an adrenal bleed (3.6 x 3.1 x 2.4 cc). The infant was followed at 2 weeks (2-dimensional size diminished to 1.5 x. 2.4 cm2 on ultrasonography) and then at 6 weeks to document that the adrenal bleed continued to involute. Urine catecholamines were normal.
-
Table. A Consensus Pretreatment Classification schema by the International Neuroblastoma Risk Group (INRG). This schema is based in the INRG stage, age, histologic category, tumor grade of differentiation, MYCN sastus, 11q-aberrations and DNA ploidy. A combination of these characteristics results in four risk groups noted in the last column: very low, low, intermediate and high risk, with the following 5 year EFS: >85%, >75%-85%, >50%-75%, and < 50%. These risk groups are distributed among the different stages and labeled alphabetically from A to R (without letters L and M to avoid confusion with the INRG stage notation). Notations in the table are as follow: L1, localized tumor confined to one body compartment; L2, locoregional tumor with presence of one or more risk factors defined radiologically; M, distant metastatic disease (except stage MS); MS, metastatic disease confined to skin, liver and/or bone marrow in children < 18 months of age. GN, ganglioneuroma; GNB, ganglioneuroblastoma; Amp, amplified; n/amp, not amplified. (Adapted from The International Neuroblastoma Risk Group (INRG) Classifications System: An INRG Task Force Report by Cohn, et al. Journal of Clinical Oncology 27(2):289-297, 2009).
Tables
Risk Group |
Stage |
Age |
MYCN Amplification Status |
Ploidy |
Shimada |
Low |
1 |
Any |
Any |
Any |
Any |
Low |
2a/2b |
Any |
Non-amp |
Any |
Any |
High |
2a/2b |
Any |
Amp |
Any |
Any |
Intermediate |
3 |
< 547d |
Non-amp |
Any |
Any |
Intermediate |
3 |
≥547d |
Non-amp |
Any |
Favorable |
High |
3 |
Any |
Amp |
Any |
Any |
High |
3 |
≥547d |
Non-amp |
Any |
Unfavorable |
High |
4 |
< 365d |
Amp |
Any |
Any |
Intermediate |
4 |
< 365d |
Non-amp |
Any |
Any |
High |
4 |
365-547d |
Amp |
Any |
Any |
High |
4 |
365-547d |
Any |
Diploid |
Any |
High |
4 |
365-547 |
Any |
Any |
Unfavorable |
Intermediate |
4 |
365-547d |
Non-amp |
Hyper |
Favorable |
High |
4 |
≥547d |
Any |
Any |
Any |
Low |
4s |
< 365d |
Non-amp |
Hyper |
Favorable |
Intermediate |
4s |
< 365d |
Non-amp |
Diploid |
Any |
Intermediate |
4s |
< 365d |
Non-amp |
Any |
Unfavorable |
High |
4s |
< 365d |
Amp |
Any |
Any |






