Background
Glycogen storage disease type V, also known as McArdle disease, is an inherited disorder of glycogen metabolism that primarily affects skeletal muscles. [1, 2, 3] It was first identified in 1951, when McArdle described a 30-year-old man who experienced muscle pain followed by weakness and stiffness after exercise. The venous lactate level of this patient failed to increase after ischemic activity. [4] In 1959, myophosphorylase was discovered, and its activity was found to be absent in individuals with McArdle disease. The typical features of McArdle disease include exercise intolerance with myalgia, early fatigue, muscle stiffness, and cramping, which are all relieved by rest. Following a short period of rest, most patients experience a “second wind” phenomenon and can resume exercise without difficulty, which is a pathognomonic feature of the disease.
About half of patients with McArdle disease experience rhabdomyolysis and myoglobinuria following vigorous exercise, and some may develop renal failure. [2, 3] Mild proximal muscle weakness occurs in approximately one third of patients and is more common in older patients. A fatal infantile form of McArdle disease, characterized by hypotonia, generalized muscle weakness, and progressive respiratory insufficiency, has been reported. In addition, a late-onset form without manifestations until the sixth decade of life has been described.
Pathophysiology
McArdle disease is caused by a deficiency of myophosphorylase (alpha-1,4-glucan orthophosphate glycosyl transferase). [1, 2] In healthy individuals, myophosphorylase initiates glycogen breakdown by removing 1,4-glucosyl groups from glycogen with the release of glucose-1-phosphate (see image below).
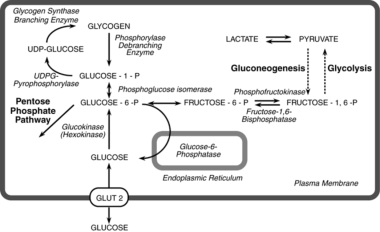 Major pathways of synthesis and breakdown of glycogen in liver. The broken line indicates that several enzymes have been omitted between pyruvate and fructose-1,6-P2. GLUT= glucose transport protein; UDP=uridine diphosphate; UDPG=uridine diphosphate-glucose. Courtesy of McGraw-Hill Education (Fig 71-2 from Valle D, Beaudet AL, Vogelstein B, et al. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. Chapter 71: Glycogen Storage Diseases. Available at: http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62672129).
Major pathways of synthesis and breakdown of glycogen in liver. The broken line indicates that several enzymes have been omitted between pyruvate and fructose-1,6-P2. GLUT= glucose transport protein; UDP=uridine diphosphate; UDPG=uridine diphosphate-glucose. Courtesy of McGraw-Hill Education (Fig 71-2 from Valle D, Beaudet AL, Vogelstein B, et al. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. Chapter 71: Glycogen Storage Diseases. Available at: http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62672129).
Several tissue-specific isoforms of phosphorylase are noted. Although myophosphorylase is present in cardiac muscle and the brain, it is the only isoform present in skeletal muscle. The liver isoform is deficient in individuals with glycogen-storage disease type VI (Hers disease). Most patients with McArdle disease have undetectable myophosphorylase activity and, thus, are unable to utilize energy (release glucose) from their glycogen stores in muscles. Rarely, patients have residual enzyme activity (< 30% of normal).
During aerobic exercise (eg, walking, gentle swimming, jogging, cycling), the skeletal muscle depends on bloodborne substrates such as free fatty acid as fuel for energy. Free fatty acids are oxidized in the mitochondrial beta-oxidation pathway to produce acetyl-CoA, which is further metabolized through the Krebs cycle and the mitochondrial respiratory chain, resulting in adenosine triphosphate (ATP) production. During anaerobic exercise (eg, weightlifting, sprinting), the myophosphorylase in the skeletal muscles converts glycogen to glucose, which enters the glycolysis pathway to produce ATP anaerobically.
Any exercise has an anaerobic component in the first few minutes. Thereafter, the duration and intensity of exercise determines the type of fuel source used by the skeletal muscles (eg, anaerobic glycolysis, blood glucose, muscle glycogen followed by aerobic glycolysis and fatty acid oxidation). The increased levels of fatty acids as additional energy sources for muscle accounts for the “second wind” phenomenon. [5, 6]
Epidemiology
Frequency
United States
McArdle disease is inherited in an autosomal recessive manner. The frequency is estimated at 1 per 100,000 population. However, only a few hundred cases have been reported. This disorder is probably underdiagnosed because of the mild symptoms in many patients. The early-onset form is extremely rare; only several cases have been reported. The late-onset form is also exceedingly rare. The gene for myophosphorylase (PGYM) is localized on chromosome 11. More than 65 mutations have been identified. Manifesting heterozygotes occur, and synergistic heterozygosity involving this gene may account for muscle symptoms in some heterozygotes.
Mortality/Morbidity
Muscular weakness and fatigue are observed. Tiredness, weakness, and cramping can interfere with normal activity. Some patients can adapt their exercise patterns to take advantage of the “second wind” phenomenon. Fixed proximal weakness occurs in as many as one third of patients. Rhabdomyolysis following vigorous exercise may result in myoglobinuria. As many as one third of patients with myoglobinuria develop acute renal failure. Death is caused by respiratory failure due to severe rapidly progressive muscular weakness.
Sex
McArdle disease is inherited in an autosomal recessive pattern. [3] The disease has been reported more often in males than in females, probably reflecting small numbers and sampling effects. Genetic data and disease severity correlations were studied in 99 patients of Spanish descent with McArdle disease; 41% of the female subjects scored in the highest severity category compared with only 20% of the males. [7, 8] In a 2017 reanalysis by the same authors, the main clinical features of the disease remained essentially unchanged compared with those found in the prior study, except for a slight decrease in the number of individuals with fixed muscle weakness (decreased from 25% to 21%). [9]
Age
McArdle disease typically presents in the second to third decade of life with limited exercise tolerance. The fatal infantile form manifests in the newborn period.
Prognosis
McArdle disease typically has a relatively benign nature when severe rhabdomyolysis is avoided. [3] Limitation or adaptation of exercise to avoid symptoms may be necessary. Acute renal failure requires appropriate treatment. Progression to chronic renal disease has not been described, but acute renal failure due to myoglobinuria is potentially life threatening.
More recent studies suggest heterogeneity of clinical severity, with 8% of patients being asymptomatic during normal daily life and 21% showing limitations during daily activities and fixed muscle weakness. [9] The evidence also supports adopting an active lifestyle, which is key in patients with McArdle disease. [9]
The current evidence also suggests that the disease does not appear to adversely affect pregnancy course or childbirth. [8] However, the optimal method of delivery (vaginal or caesarean) has not been determined. [9]
Patient Education
Educate patients about modifying their activity in order to prevent rhabdomyolysis. Patients should avoid extreme isometric exercise. [3]
Educate patients about the “second wind” phenomenon. [10]
Genetic counseling should also be provided to educate parents and affected individuals about recurrence risk for future pregnancies. [3]
-
Enzyme histochemistry of 19-year-old male with McArdle disease.
-
Major pathways of synthesis and breakdown of glycogen in liver. The broken line indicates that several enzymes have been omitted between pyruvate and fructose-1,6-P2. GLUT= glucose transport protein; UDP=uridine diphosphate; UDPG=uridine diphosphate-glucose. Courtesy of McGraw-Hill Education (Fig 71-2 from Valle D, Beaudet AL, Vogelstein B, et al. The Online Metabolic and Molecular Bases of Inherited Disease. 2014. Chapter 71: Glycogen Storage Diseases. Available at: http://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62672129).








