Background
The definition and classification of hypertrophic cardiomyopathy (HCM) have varied over the decades, primarily because the phenotypic expression of ventricular hypertrophy can result from a myriad of diseases, especially among children. In the past, this disease entity has been called idiopathic hypertrophic subaortic stenosis (IHSS), asymmetric septal hypertrophy (ASH), dynamic muscular subaortic stenosis, diffuse muscular subaortic stenosis, hypertrophic subaortic stenosis, Teare disease, Brock disease, and hypertrophic obstructive cardiomyopathy (HOCM).
At present, most authorities agree to call this disease entity "hypertrophic cardiomyopathy," which is then subdivided into obstructive and nonobstructive types, depending upon the presence of left ventricular outflow tract obstruction. For the purposes of this article, HCM is a primary cardiac disorder that results from known or suspected genetic defects in sarcomeric proteins of the cardiac myocyte. The disorder is thought to be inherited in an autosomal dominant fashion with variable penetrance and variable expressivity.
The hallmark of HCM is myocardial hypertrophy that is inappropriate and often asymmetric and that occurs in the absence of an obvious inciting hypertrophic stimulus. Although any region of the left ventricle can be affected, hypertrophy frequently involves the interventricular septum, which can result in outflow tract obstruction. Patients typically have preserved systolic function with impaired left ventricular compliance that results in diastolic dysfunction, whether or not outflow tract obstruction is present.
HCM has a complex set of symptoms and potentially devastating consequences for patients and their families. The clinical presentation and course vary widely; some children are completely asymptomatic, whereas others experience sudden cardiac death (see Presentation.) In fact, among adolescent children, HCM is the leading cause of sudden cardiac death during exertion.
HCM is a chronic illness that imposes lifestyle restrictions. Management of pediatric HCM patients involves long-term care and close observation (especially during puberty), medical or surgical treatment for symptoms, identification and treatment of those at risk for sudden death, and screening of other at-risk family members. [1, 2] (See Treatment.)
Pathophysiology
Defects in genes that encode for the sarcomeric proteins (eg, myosin heavy chain, actin, tropomyosin, titin) provide the molecular basis for most cases of familial hypertrophic cardiomyopathy (HCM). These defects result in myofibril disarray and fibrosis that progress over time and contribute to ventricular hypertrophy. The chaotic cellular architecture occurs even in areas of the myocardium that are not hypertrophied and may be arrhythmogenic substrates for ventricular tachycardia or ventricular fibrillation.
Patients with HCM also have abnormal intramural coronary arteries, with thickened intima leading to vessel narrowing and possible inability to supply the oxygen demand of the hypertrophied myocardium; ischemia, cell death, and scar formation result.
Although the ventricle becomes hypertrophic, the ventricular cavity itself does not dilate, remaining normal or even small in size. The contractile (systolic) function of the ventricle remains intact; however, impaired relaxation and filling often occur. The impaired ventricular compliance and diastolic dysfunction lead to elevated end-diastolic pressures.
In late stages of the disease, patients may progress to heart failure with ventricular dilatation. Studies in mice with a positive HCM genotype but a negative phenotype suggest that administration of the calcium channel blocker diltiazem before the development of ventricular hypertrophy may prevent disease in this animal model. [3] Studies of calcium channel blocker therapy in presymptomatic humans are currently being conducted.
Since the initial descriptions of HCM, the feature that has attracted greatest attention is the dynamic pressure gradient across the left ventricular outflow tract. The pressure gradient appears to be related to several factors, including hypertrophy of the interventricular septum into the outflow tract, possible abnormalities in location of the mitral valve apparatus, and systolic anterior motion of the mitral valve against the hypertrophied septum.
The degree of obstruction varies among patients. Some patients have no gradient, whereas others develop obstruction only with exertion. The obstruction is also dynamic and depends on the patient’s volume status; volume depletion increases the outflow gradient, whereas volume repletion decreases obstruction. The degree of obstruction does not correlate with the risk of sudden cardiac death.
Etiology
In 1989, Jarcho et al reported the genetic basis for hypertrophic cardiomyopathy (HCM) and the existence of a disease gene located on the long arm of chromosome 14, which was subsequently found to encode for the beta cardiac myosin heavy chain. [4] At least 15 different genes on at least 6 chromosomes are associated with HCM, and more than 400 different, predominantly missense, mutations have been discovered. These genes encode for sarcomeric proteins such as myosin heavy chain, actin, titin, myosin-binding protein, tropomyosin, and others (see the image below). An estimated 60% of HCM cases carry mutations in 1 of 8 sarcomere protein genes, primarily variants of nonsense MYBPC3 and missense MYH7. [5]
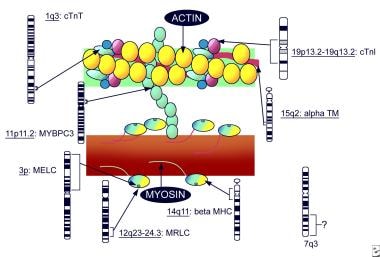 Pediatric Hypertrophic Cardiomyopathy. Sarcomeric genes involved in hypertrophic cardiomyopathy (adapted from Priori 1999).
Pediatric Hypertrophic Cardiomyopathy. Sarcomeric genes involved in hypertrophic cardiomyopathy (adapted from Priori 1999).
Familial HCM occurs as an autosomal dominant inherited disease in approximately 50% of individuals with the disorder. The variable penetrance and expression of disease among family members carrying the same genetic defect is explained by certain individual modifier genes that affect presentation. Some, if not all, of the sporadic forms of the disease may be due to spontaneous mutations.
Genetic testing for HCM is commercially available. Among patients who are clinically diagnosed and undergo genetic testing, 50-80% have a positive test result. This suggests that novel HCM mutations are yet to be discovered.
In children, the phenotypic expression of ventricular hypertrophy can occur secondary to other pediatric diseases that should be differentiated from HCM. These include inborn errors of metabolism (eg, Pompe disease and Barth syndrome), malformation syndromes (eg, Noonan syndrome), and neuromuscular disorders (eg, Friedrich ataxia, Duchenne muscular dystrophy). [6]
Two specific glycogen-storage disorders can also lead to familial HCM and involve defects in protein kinase gamma-2 (PRKAG2), which result in a familial glycogen-accumulation cardiomyopathy associated with Wolff-Parkinson-White syndrome, and defects in lysosomal-associated membrane protein 2 (LAMP2), which result in Danon disease. Ventricular hypertrophy secondary to athletic conditioning—the so-called athlete’s heart—should also be considered in the differential diagnosis.
Epidemiology
United States statistics
Hypertrophic cardiomyopathy (HCM) is relatively common in the United States, with an estimated prevalence of 0.2% (1 case per 500 population [7] ) in adults. Studies among children suggest a lower incidence for disease expression beginning in childhood, with a rate of 3-5 cases per 1 million children. Morphologic evidence of disease is found on echocardiography in approximately 25% of first-degree relatives of patients with HCM, a finding consistent with variable expressivity.
International statistics
The prevalence of hypertrophic cardiomyopathy is thought to be similar throughout the world; however, certain variants, such as apical hypertrophy, may be more predominant among Asians.
Race-, age-, and sex-related demographics
HCM does not have a racial or ethnic predisposition and has been reported in patients of all races.
This condition may occur at any age, from the newborn to the elderly. Overall, its most common presentation is in the third decade of life. Among children younger than 18 years diagnosed with HCM, the median age at diagnosis is 7 years; one third are diagnosed before age 1 year.
The genetic inheritance pattern is autosomal dominant, without any sex predilection. Although no sex difference is noted among infants diagnosed with HCM before age 1 year, among children diagnosed after age 1 year, HCM is more commonly identified in males than in females. Modifying genetic, hormonal, and environmental factors may lead to higher likelihood of identification, more apparent symptoms, or higher degrees of left ventricular outflow obstruction in males, thus resulting in more prominent physical examination findings.
Prognosis
Studies among children with hypertrophic cardiomyopathy (HCM) suggest that mortality is lower than previously reported, probably because disease recognition has improved, allowing diagnosis of patients with less severe disease. [8] The overall mortality is approximately 1% per year. Infants diagnosed with HCM before age 1 year appear to have the highest mortality rates; a 5 year survival rate of ~70% was found in the Pediatric Cardiomyopathy Registry. [8] Infants who survive beyond age 1 year or children diagnosed after age 1 year have an overall mortality of 1% per year. [9]
Sudden death is the most common cause of death in children with HCM. A subgroup of children appear to have a higher risk of sudden cardiac death, reportedly as high as 4-6%. Given the overall mortality of 1% per year, it appears that children not in this higher-risk subgroup have very low mortalities, with a possible normal life expectancy. A recent study demonstrated that 25% of HCM patients, detected by school screening, exhibited life-threatening events during 15-year follow-up. [10]
Complications
Complications of HCM may include the following:
-
Congestive heart failure
-
Arrhythmia
-
Infective mitral endocarditis
-
Atrial fibrillation with mural thrombosis formation
-
Sudden death
Patient Education
Individuals with hypertrophic cardiomyopathy (HCM) should be advised to avoid strenuous activity, anaerobic exercise (eg, weightlifting), and high-level competitive sports. Activity restrictions should be imposed. [7]
Family members of persons with HCM should learn cardiopulmonary resuscitation (CPR). Both the patient and the family members should be referred for psychosocial counseling.
-
Pediatric Hypertrophic Cardiomyopathy. Hypertrophic cardiomyopathy. Image courtesy of Michael E. Zevitz, MD
-
Pediatric Hypertrophic Cardiomyopathy. Sarcomeric genes involved in hypertrophic cardiomyopathy (adapted from Priori 1999).
-
Pediatric Hypertrophic Cardiomyopathy. ECG of a 16-year-old with hypertrophic cardiomyopathy (HCM), demonstrating left ventricular hypertrophy pattern and "pseudo-preexcitation."





