Practice Essentials
Hyperparathyroidism refers to a serum parathyroid hormone (PTH) level above the normal range. PTH secretion is regulated by the action of serum ionized calcium on the calcium sensing receptor (CASR) in parathyroid chief cells. Physiologically, calcium level is maintained within normal limits mainly due to this calcium sensing mechanism. Hyperparathyroidism may be classified into three major subtypes based on pathophysiology: primary, secondary, and tertiary hyperparathyroidism.
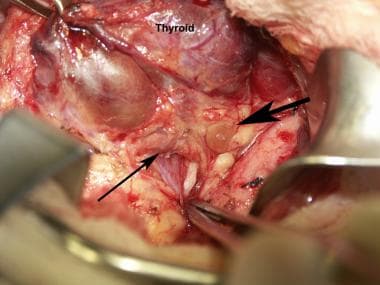 Normal parathyroid glands as seen during a thyroidectomy. The large arrow points to the superior parathyroid. The thinner arrow points to the inferior parathyroid. The forceps points toward the recurrent laryngeal nerve. The patient's head is toward the right.
Normal parathyroid glands as seen during a thyroidectomy. The large arrow points to the superior parathyroid. The thinner arrow points to the inferior parathyroid. The forceps points toward the recurrent laryngeal nerve. The patient's head is toward the right.
Signs and symptoms of hypercalcemia in primary or tertiary hyperparathyroidism
Clinical features include the following:
-
Depressed mood, anxiety, and altered mental status
-
Poor weight gain and failure to thrive in younger children and infants
-
Bradycardia, with or without irregular heartbeat
-
Signs of dehydration, such as dry mucous membranes, prolonged capillary refill, tenting of skin
-
Abdominal pain due to increased gastric acid secretion, constipation, peptic ulcers, or acute pancreatitis
-
Hypotonia
-
Flank pain due to renal calculi
-
Signs of fractures such as bone pain and deformities are more common in neonates with severe neonatal hyperparathyroidism
See Presentation for more detail.
Diagnosis
Laboratory studies
Serum biochemistry studies include the following:
-
Parathyroid hormone (PTH) level
-
Serum calcium, total or ionized calcium
-
Serum albumin
-
Serum phosphorus
-
25 OH Vitamin D level
Genetic studies
Urine studies: A calcium/creatinine ratio from a spot urine sample
Biochemical markers of bone turnover include the following:
-
Serum levels of osteocalcin or bone-specific alkaline phosphatase
-
Serum C-telopeptide of type I collagen and Urinary N-telopeptide (NTx)
Genetic tests for familial forms of primary hyperparathyroidism
Imaging studies
Imaging studies used in the workup include the following:
-
Sestamibi scanning paired with single-photon emission computerized tomography
-
Bone densitometry, determined by dual energy x-ray absorptiometry
-
Renal ultrasound
-
Skeletal radiographs
See Workup for more detail.
Management
Familial hypocalciuric hypercalcemia is mild, usually asymptomatic, and therefore does not require specific therapy. Treatment for severe neonatal hyperparathyroidism has been surgical but the development of new drugs has allowed for successful medical management in several cases. These therapies are considered experimental and include calcimimetics (cinacalcet) and bisphosphonates (pamidronate), which have been used as monotherapy or as combination therapy.
Treatment of primary hyperparathyroidism due to an adenoma or multiglandular disease is surgical.
See Treatment and Medication for more detail.
Pathophysiology
Primary hyperparathyroidism (PHPT) entails an abnormality in parathyroid cell function leading to hypercalcemia with an inappropriately normal or elevated PTH level. The etiology and pathology of PHPT is very different in neonates and older children. Most neonatal cases are due to inactivating mutations of the calcium sensing receptor (CASR) causing severe neonatal hyperparathyroidism (NSHPT). On the other hand, in children and adolescents PHPT is most frequently (80-92%) due to a single benign parathyroid adenoma, and less commonly due to multiglandular disease (MGD). [1, 2, 3, 4, 5, 6, 7, 8] MGD is more commonly observed in the multiple endocrine neoplasia syndromes MEN1, MEN2A, and MEN4 or as part of the hyperparathyroidism jaw tumor syndrome (HPT-JT). Germline mutations in several genes have been identified in MGD as well as in non-syndromic familial isolated hyperparathyroidism. Somatic mutations in different genes have been related to a minority of cases of sporadic parathyroid adenomas. [9, 10] The differential diagnosis for primary hyperparathyroidism includes a usually asymptomatic form of primary hyperparathyroidism due to heterozygous inactivating CASR mutations, familial hypocalciuric hypercalcemia (FHH). Parathyroid carcinoma is very rare in adults and children(< 1%). [11]
A meta-analysis by Roizen et al concluded that hypercalcemia and hypercalciuria is greater in juvenile primary hyperparathyroidism than adult primary hyperparathyroidism with serum intact PTH at similar concentrations which suggested a different pathophysiology between pediatric and adult cases. [12]
Secondary hyperparathyroidism refers to an elevated PTH level in the context of low or normal serum calcium levels. This disorder may be caused by hyperphosphatemia as observed in chronic renal failure, or by hypocalcemia as in malabsorption or vitamin D deficiency. The elevated PTH level in these cases reflects a normal response to a stimulus, and it normalizes by treating the underlying pathology.
Tertiary hyperparathyroidism occurs when parathyroid hyperplasia becomes so severe that removal of the underlying cause does not eliminate the stimulus for PTH secretion and hypertrophic chief cells become autonomous. This usually presents as the progression of chronic secondary hyperparathyroidism
This chapter will focus on primary hyperparathyroidism. In these cases, inappropriately elevated parathyroid hormone secretion leads to hypercalcemia and hypophosphatemia. PTH increases renal calcium reabsorption at the distal convoluted tubule and increases intestinal calcium absorption indirectly by increasing the production of 1,25 (OH)2 vitamin D by stimulating the 1 α hydroxylation of 25 OH vitamin D in the proximal renal tubules. 1,25 (OH)2 Vitamin D in turn stimulates active intestinal calcium transport. PTH also leads to hypercalcemia by increasing bone resorption. PTH indirectly stimulates bone resorption by acting on the osteoblast PTH receptor, which then signals the osteoclast to produce various substances, among them is the ligand of the receptor activator of the nuclear transcription factor NF-kappa B (RANK), known as RANK ligand (RANKL), which can stimulate osteoclast differentiation and proliferation. PTH leads to hypophosphatemia by decreasing renal phosphate reabsorption.
Etiology
The main causes of hyperparathyroidism by age groups are detailed in Table 1. Homozygous inactivating CASR mutations lead to NSHPT, while heterozygous mutations are observed mainly in the milder form, familial hypocalciuric hypercalcemia (FHH), although cases of NSHPT with heterozygous mutations have been reported. [13] FHH is inherited in an autosomal dominant manner, but may be sporadic. Additionally, mutations in the G protein subunit α 1 and AP2S1 gene have recently been found to cause FHH. [14, 15] FHH manifests with mild hypercalcemia associated with hypocalciuria, most often not requiring specific treatment.
In older children and adolescents primary hyperparathyroidism is most often caused by isolated sporadic parathyroid adenomas (80-92%). [16] The remaining are due to multiglandular parathyroid hyperplasia observed in familial endocrine neoplastic syndromes, in which cases a family history of endocrine tumors with an autosomal dominant pattern of inheritance often exists. Multiple endocrine neoplasia type 1 is caused by a mutation in the gene MEN1 which encodes menin, a tumor suppressor protein. Hyperparathyroidism is the presenting sign in 95% of patients with MEN1, while pancreatic and pituitary tumors develop in 40 and 30% of cases respectively. [9] MEN 2A is caused by mutations in the c-ret proto-oncogene (RET), which encodes a tyrosine kinase receptor. MEN 2A presents with medullary thyroid carcinoma associated with hyperparathyroidism (20%) and pheochromocytoma (50%). MEN 4 is caused by mutations in CDKN1B, a cyclin dependent kinase inhibitor, and has been diagnosed in a small percentage of patients with the tumors associated with MEN1 in addition to adrenal, gonadal and thyroid tumors. Hyperparathyroidism jaw tumor syndrome is caused by mutations in cell division cycle 73 gene (CDC73), which codes for parafibromin, a nuclear protein thought to be a tumor suppressor gene. In this syndrome, osseous fibromas of the jaw are associated with parathyroid adenomas or carcinomas, and renal and uterine tumors may also be found. [9] In general, parathyroid carcinoma is very rare in children with 11 cases reported in the English literature in the past 41 years. [11]
Table 1. Causes of hyperparathyroidism (Open Table in a new window)
Cause |
Gene |
OMIM |
1. Neonate/Infant |
||
|
||
- Familial hypocalciuric hypercalcemia |
Heterozygous inactivating CASR mutations |
145980 |
- Neonatal severe hyperparathyroidism |
Homozygous inactivating CASR mutations |
239200 |
|
||
- Maternal hypoparathyroidism |
||
- Maternal pseudohypoparathyroidism |
||
- Maternal Vitamin D deficiency |
||
2. Child/adolescent |
||
|
||
- Sporadic adenomas |
||
- Familial |
||
* Familial hypocalciuric hypercalcemia (FHH) |
CASR |
145980 |
* Multiple Endocrine Neoplasia type 1 (MEN1) |
MEN1 |
131100 |
* Multiple Endocrine Neoplasia type 2a |
RET |
171400 |
* Multiple Endocrine Neoplasia type 4 |
CDKN1B |
|
* Familial hyperparathyroidism jaw tumor syndrome (HRPT2) |
CDC73 | 145001 |
|
||
- Autoantibodies to the CASR |
||
|
||
- Renal failure/post renal transplant |
||
- Chronic hyperphosphatemia |
||
- Vitamin D deficiency |
||
* Nutritional |
||
* Malabsorption |
||
-Vitamin D dependent rickets type 1 VDDR1 |
CYP27B1 | 264700 |
|
|
|
- Vitamin D dependent rickets type 2 VDDR2 |
Vitamin D receptor | 277440 |
|
|
|
-Other causes of hypocalcemia |
||
Poor intake Drugs |
Epidemiology
The estimated incidence of primary hyperparathyroidism (PHPT) in pediatric patients is 1 per 200-300,000 and its prevalence is 2-5 in 100,000. [1, 6] It has a higher predominance in adolescents, but its incidence is still much lower in this population than in adults where it has been estimated at 1:500-2000. [17] In adults, PHPT is more frequent in females, while most pediatric series find no difference in distribution by sex. [4]
Prognosis
Prognosis depends on the etiology. For primary hyperparathyroidism due to a parathyroid adenoma, parathyroidectomy should be curative if the condition occurs in isolation. However, if it is associated with other tumors, prognosis would depend on the management of accompanying tumors. NSHPT has a high mortality rate if untreated. Secondary hyperparathyroidism is cured by treating the underlying pathology.
Complications
Acute complications if untreated are those of hypercalcemia.
Chronic complications include the following:
-
Failure to thrive
-
Low bone mineral density
-
Fractures
-
Nephrolithiasis or nephrocalcinosis
-
Renal failure
Surgical complications include recurrent laryngeal nerve injury, bleeding, and infection. Complications of primary hyperparathyroidism include consequences of hypercalcemia, such as nephrolithiasis, dehydration, and cardiac arrhythmias, and complications due to end organ involvement such as renal failure and fractures.
Patient Education
Patients with primary hyperparathyroidism must understand the following:
-
Location and function of parathyroid gland and PTH
-
Effects of hypercalcemia on the body (eg, dehydration, neurological symptoms, arrhythmia, stones, bone demineralization, increased fracture risk)
-
Lack of success in managing most cases of primary hyperparathyroidism medically, need for surgical consultation, and resection of one or more parathyroid glands
-
Normal parathyroid glands as seen during a thyroidectomy. The large arrow points to the superior parathyroid. The thinner arrow points to the inferior parathyroid. The forceps points toward the recurrent laryngeal nerve. The patient's head is toward the right.
Tables
Cause |
Gene |
OMIM |
1. Neonate/Infant |
||
|
||
- Familial hypocalciuric hypercalcemia |
Heterozygous inactivating CASR mutations |
145980 |
- Neonatal severe hyperparathyroidism |
Homozygous inactivating CASR mutations |
239200 |
|
||
- Maternal hypoparathyroidism |
||
- Maternal pseudohypoparathyroidism |
||
- Maternal Vitamin D deficiency |
||
2. Child/adolescent |
||
|
||
- Sporadic adenomas |
||
- Familial |
||
* Familial hypocalciuric hypercalcemia (FHH) |
CASR |
145980 |
* Multiple Endocrine Neoplasia type 1 (MEN1) |
MEN1 |
131100 |
* Multiple Endocrine Neoplasia type 2a |
RET |
171400 |
* Multiple Endocrine Neoplasia type 4 |
CDKN1B |
|
* Familial hyperparathyroidism jaw tumor syndrome (HRPT2) |
CDC73 | 145001 |
|
||
- Autoantibodies to the CASR |
||
|
||
- Renal failure/post renal transplant |
||
- Chronic hyperphosphatemia |
||
- Vitamin D deficiency |
||
* Nutritional |
||
* Malabsorption |
||
-Vitamin D dependent rickets type 1 VDDR1 |
CYP27B1 | 264700 |
|
|
|
- Vitamin D dependent rickets type 2 VDDR2 |
Vitamin D receptor | 277440 |
|
|
|
-Other causes of hypocalcemia |
||
Poor intake Drugs |