Practice Essentials
Moyamoya disease is a progressive, occlusive disease of the cerebral vasculature with particular involvement of the circle of Willis and the arteries that feed it. [1]
Signs and symptoms
Children and adults with moyamoya disease may have different clinical presentations. Adults experience hemorrhage more commonly; cerebral ischemic events are more common in children.
Diagnosis
Cerebral angiography is the criterion standard for the diagnosis of moyamoya disease. The following findings support the diagnosis:
-
Stenosis or occlusion at the terminal portion of the internal carotid artery or the proximal portion of the anterior or middle cerebral arteries
-
Abnormal vascular networks in the vicinity of the occlusive or stenotic areas
-
Bilaterality of the described findings (although some patients may present with unilateral involvement and then progress)
Management
Various surgical procedures have been used in the treatment of moyamoya disease, with the goal of revascularizing the ischemic hemisphere, including the following: [2]
-
Superficial temporal artery–middle cerebral artery (STA-MCA) anastomosis
-
Encephaloduroarteriosynangiosis (EDAS) [3]
-
Encephaloduroarteriomyosynangiosis (EDAMS)
-
Pial synangiosis
-
Omental transplantation
Background
Moyamoya disease is a progressive, occlusive disease of the cerebral vasculature with particular involvement of the circle of Willis and the arteries that feed it. [1] The image below is a schematic representation of the circle of Willis, the arteries of the brain, and the brainstem. (See Etiology.)
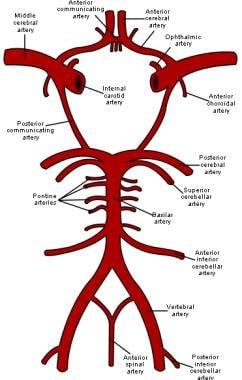 Schematic representation of the circle of Willis, arteries of the brain, and brain stem.
Schematic representation of the circle of Willis, arteries of the brain, and brain stem.
The term moyamoya (Japanese for "puff of smoke") refers to the appearance on angiography of abnormal vascular collateral networks that develop adjacent to the stenotic vessels. The steno-occlusive areas are usually bilateral, but unilateral involvement does not exclude the diagnosis. (See Workup.)
Blood vessel walls consist of 3 layers: the intima is the innermost layer; the media is a muscular middle layer; and the adventitia is the outermost layer. Separating the intima and media is the internal elastic lamina, an elastic membrane that is considered the outermost part of the intima. Pathologically, moyamoya disease is characterized by intimal thickening in the walls of the terminal portions of the internal carotid vessels bilaterally. The proliferating intima may contain lipid deposits. The anterior, middle, and posterior cerebral arteries that emanate from the circle of Willis may show varying degrees of stenosis or occlusion. This is associated with fibrocellular thickening of the intima, waving of the internal elastic lamina, and thinning of the media. (See Etiology, Workup, Treatment, and Medication.)
Numerous small vascular channels can be seen around the circle of Willis. These are perforators and anastomotic branches. The pia mater may also have reticular conglomerates of small vessels.
Etiology
The cause of moyamoya disease is not known. The disease is believed to be genetic. Fukui reported a family history in 10% of patients with the disorder. Moreover, Mineharu suggested that familial moyamoya disease is autosomal dominant with incomplete penetrance that depends on age and genomic imprinting factors. [4] Genetically, susceptibility loci have been found on 3p, 6p, 17q, and band 8q23. Mineharu et al have found a specific gene locus, q25.3, on chromosome 17. [5] A genome-wide association study identified RNF213 as the first gene associated with moyamoya. [6] One meta-analysis demonstrated that there are strong associations between p.R4859K and p.R4810K polymorphisms of the RNF213 gene and moyamoya disease. [7]
People with moyamoya disease have been found to have a higher incidence of elevated thyroid antibodies. [8] While this is an association in some individuals, the significance is not clear. However, it suggests that immune abnormalities and several others as listed below may play some role in moyamoya disease.
Associated diseases
Although moyamoya disease may occur by itself in a previously healthy individual, many disease states have been reported in association with moyamoya disease, including the following:
-
Immunologic [9, 10] - Graves disease/thyrotoxicosis, systemic lupus erythematosus
-
Infections [11] - Leptospirosis, tuberculosis, varicella
-
Vascular diseases [18] - Atherosclerotic disease, arteriovenous malformation, coarctation of the aorta and fibromuscular dysplasia, cranial trauma, radiation injury, parasellar tumors, and hypertension
These associated conditions may not be causative, but they do warrant consideration due to their impact on treatment. In the presence of these risk factors, the condition is referred to as moyamoya syndrome.
Epidemiology
A study indicated that the prevalence of moyamoya disease in California and Washington was 0.086 case per 100,000 people. [19] In this study, the breakdown based on ethnicity as ratio to whites was 4.6 for Asian Americans, 2.2 for African Americans, and 0.5 for Hispanics.
The incidence of moyamoya disease is highest in Japan. [20] The prevalence and incidence of the disorder there has been reported to be 3.16 cases and 0.35 case per 100,000 people, respectively.
Race-, sex-, and age-related demographics
Moyamoya disease occurs primarily in Asians but can also occur (with varying degrees of severity) in whites, blacks, Haitians, and Hispanics.
The female-to-male ratio of moyamoya disease is 1.8:1. Ages for patients with moyamoya disease range from 6 months to 67 years, with the highest peak in the first decade and smaller peaks in the third and fourth decades. [20]
Prognosis
Death from moyamoya disease is usually from hemorrhage. The outcome of the disease depends on the severity and nature of the hemorrhage; the prognosis depends on recurrent attacks.
Mortality rates from moyamoya disease are approximately 10% in adults and 4.3% in children. About 50–60% of affected individuals experience a gradual deterioration of cognitive function, presumably from recurrent strokes.
Patients with moyamoya disease who present for treatment while symptoms are evolving have a better prognosis than do those who present with static symptoms (which probably indicate a completed stroke).
There is still lots of work to be done, as there are still many unresolved issues about moyamoya disease and syndrome, including lack of clear diagnostic criteria, particular biomarkers, precise understanding of the underlying pathophysiology, and stronger evidence for treatment guidelines. [21]
-
Schematic representation of the circle of Willis, arteries of the brain, and brain stem.



