Practice Essentials
Central neurofibromatosis, or neurofibromatosis type 2 (NF2), is a genetic disorder marked by the predisposition to develop a variety of tumors of the central and peripheral nervous systems. In contrast to neurofibromatosis type 1 (NF1), NF2 produces less frequent and usually less prominent cutaneous manifestations. (See the image below.)
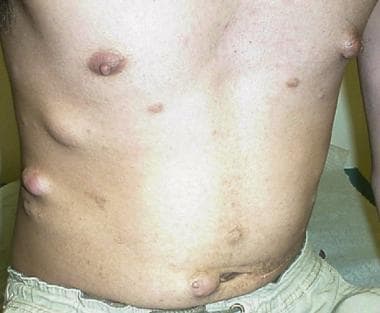 Subcutaneous and cutaneous lesions in a young man with neurofibromatosis type 2; note paucity of cafe-au-lait spots.
Subcutaneous and cutaneous lesions in a young man with neurofibromatosis type 2; note paucity of cafe-au-lait spots.
Signs and symptoms
Clinical diagnosis of NF2 requires that an individual present with at least 1 of the following clinical scenarios [1] :
-
Bilateral vestibular schwannomas
-
A first degree relative with NF2 AND
Unilateral vestibular schwannoma OR
Any two of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
-
Unilateral vestibular schwannoma AND
Any two of: meningioma, schwannoma, glioma, neurofibroma, posterior subcapsular lenticular opacities
-
Multiple meningiomas AND
Unilateral vestibular schwannoma OR
Any two of: schwannoma, glioma, neurofibroma, cataract
However, because approximately half of cases result from new mutations, family history is often negative.
Unlike NF1, which frequently is associated with a number of cutaneous diagnostic clues, NF2 is accompanied by fewer and less prominent external cutaneous signs. Presenting symptoms include the following:
-
Hearing loss, ringing in the ears, and balance problems associated with vestibular nerve lesions
-
Visual deficits
-
Cranial nerve palsies
See Clinical Presentation for more detail.
Diagnosis
Diagnosis of NF2 involves the following:
-
Genetic testing
-
Imaging studies
-
Auditory, ophthalmic, and histologic examinations
Genetic testing
-
Once the clinical diagnosis has been established in a given individual, direct molecular analysis can be offered
-
Detection rates for molecular-based testing approaches approaches 72% in simplex cases and 93% in familial cases [1]
-
If a mutation is found, other asymptomatic family members might benefit from presymptomatic testing
-
Molecular testing of tumor tissue may augment traditional molecular studies when analysis of DNA obtained from blood lymphocytes is nondiagnostic
-
For families in which no mutation can be identified in a known affected individual, linkage analysis or indirect genetic testing methods may be utilized
-
Presymptomatic testing of at-risk family members requires a vigorous informed consent process and might best be done during a genetic counseling session at a cancer, genetic, or neurofibromatosis center that specializes in such matters
Prenatal testing for NF2 is as follows:
-
When a parent has NF2, prenatal testing can be done on amniocytes or chorionic villi, either through direct gene mutation analysis when such a change has been identified or through linkage analysis
-
Prenatal testing may not be possible if the affected parent is the first affected person in the family and a mutation cannot be found
-
If a prospective parent has a known NF2 mutation, preimplantation genetic diagnosis may be possible if the couple is willing to undergo in vitro fertilization with transfer of unaffected embryos
Magnetic resonance imaging
-
MRI remains the mainstay for diagnosis and screening of CNS, cranial nerve, and spinal cord tumors
-
At-risk individuals may be monitored for CNS tumors beginning in their teens, with annual MRI scans of the head performed through their late 50s
-
MRI using 3-dimensional (3D) volumetrics is now the preferred method for following vestibular schwannoma growth over time [2]
-
Routine MRI imaging of the spinal cord probably is not indicated for asymptomatic affected or at-risk individuals
-
MRI of the spine is indicated diagnostically when an individual presents with motor or sensory changes suggestive of a spinal cord lesion or lesions
Auditory Evaluation
-
Hearing evaluations, including brainstem auditory-evoked response (BAER) may demonstrate latency abnormalities before a mass is detectable on MRI
-
Auditory screening on an annual basis may be quite useful in asymptomatic or presymptomatic individuals
-
Once a vestibular schwannoma is identified, full audiometry testing, including acoustic reflex testing as well as BAER, is useful as a means of monitoring disease progression
-
The size of the vestibular tumor often does not correlate with the degree of hearing loss
Ophthalmic Examination
-
Annual eye examinations are recommended for children and adults with NF2
-
Dilated eye examinations for lens opacities, retinal hamartomas, or epiretinal membranes may be quite useful even in a child at risk for NF2
-
Juvenile cataracts may be seen long before a child shows any evidence of vestibular schwannomas
See Workup for more detail.
Management
Medical care for patients with NF2 consists of routine examinations focusing on early detection of some of the potential complications related to CNS or spinal cord lesions. [3] Management by a team of specialists through a multidisciplinary clinic may provide the most comprehensive and cost-effective care over time. The following is an outline of reasonable guidelines in the care of the patient with NF2:
-
Annual neurologic examination looking for subtle deficits or changes in neurologic status that might suggest disease progression
-
Annual hearing screening with BAER, with referral to an audiologist for amplification, augmentation, or speech therapy recommendations
-
Annual MRI to monitor existing lesions or look for presymptomatic lesions
-
Annual ophthalmologic evaluations to monitor visual acuity
Treatment of symptomatic tumors is as follows:
-
Surgical resection represents the most common approach to clinically significant lesions
-
Rarely, radiation and/or chemotherapy may be recommended to treat disabling ependymomas
-
Palliative chemotherapy for surgically unresectable ependymomas may be attempted with lomustine, vincristine, and prednisone, or carboplatin and vincristine, following radiation therapy
-
Bevacizumab may be recommended to radiographically reduce the size of vestibular schwannomas or prolong tumor stability in patients without surgical options and can improve hearing or prolong time to hearing loss [4]
-
Lapatinib may also be considered for use in reducing the size of vestibular schwannomas and to improve hearing [4]
Tumor resection and radiotherapy
-
For small vestibular schwannomas, surgical resection and stereotactic radiosurgery may preserve hearing and facial nerve function in selected patients [5]
-
Larger tumors may require surgical resection despite irreversible hearing loss
-
A debulking procedure may result in preservation of hearing or, at the minimum, prolongation of auditory decompensation
-
Nonvestibular cranial nerve schwannomas may be treated effectively with a combination of microsurgery and radiosurgery [6]
-
Unlike vestibular lesions, intracranial meningiomas may be quite slow growing; surgical resection should be considered only when such lesions are causing serious, disabling symptoms
-
Resection of spinal cord tumors is often quite difficult, and the risks and benefits of surgery must be considered on an individual basis
-
Resection of cutaneous or subcutaneous growths can be accomplished by any competent surgeon
Auditory brainstem implants
-
Auditory brainstem implants (ABIs) have been used successfully in some patients with hearing loss secondary to vestibular schwannomas
-
ABIs often improve the patient's ability to appreciate environmental sounds and facilitate communication [7]
-
ABIs in patients with NF2 do not enable high levels of speech recognition
See Treatment and Medication for more detail.
Background
Central neurofibromatosis, or neurofibromatosis type 2 (NF2), is a multisystem genetic disorder associated with bilateral vestibular schwannomas, spinal cord schwannomas, meningiomas, gliomas, and juvenile cataracts, with a paucity of cutaneous features (which are seen more consistently in neurofibromatosis type 1 [NF1]). (See the image below.)
Subcutaneous and cutaneous lesions in a young man with neurofibromatosis type 2; note paucity of cafe-au-lait spots.
Although quite variable in its age of onset and severity of symptoms in affected individuals, NF2 is associated with significant morbidity and decreased life span. Furthermore, diagnosis in childhood is often difficult because of the absence of central nervous system (CNS) involvement at a young age. [8] (See Prognosis, Clinical, and Workup.)
Complications of NF2 may include the following (See Clinical, Workup, and Treatment):
-
Unilateral or, frequently, bilateral vestibular schwannomas leading to tinnitus, hearing loss, and/or problems with balance
-
Meningiomas, gliomas, ependymomas, and other cerebral, cerebellar, or spinal cord lesions that may result in neurologic deficits, seizures, and/or hydrocephalus
-
Peripheral nerve schwannomas, mixed tumors, and, occasionally, neurofibromas
-
Peripheral neuropathies
-
Visually significant juvenile cataracts
Etiology
Neurofibromatosis type 2 (NF2) is inherited as an autosomal dominant condition, although half of affected individuals have NF2 as a result of a new (de novo) gene mutation. The manifestations of NF2 result from mutations in (or, rarely, deletion of) the NF2 gene, located on the long arm of chromosome 22. Affected individuals need only 1 mutated or deleted NF2 gene to exhibit signs of the condition.
The NF2 gene product known as merlin serves as a tumor suppressor; decreased function or production of this protein results in a predisposition to develop a variety of tumors of the central and peripheral nervous systems. [9]
Increasing evidence indicates that merlin is involved in a number of cellular pathways and works in concert with other proteins to promote cellular adhesion and responses via the growth factor receptor. [10] Understanding these interactions may eventually lead to more effective targeted treatment strategies, since the benign nature of NF2 lesions makes tumors frequently less responsive to chemotherapy or radiation therapy.
Numerous mutations in the NF2 gene have been identified, most of which are predicted to result in production of a truncated protein with loss of its usual function.
Epidemiology
The estimated incidence of neurofibromatosis type 2 (NF2) is 1 in 37,000 per year, with about half of affected individuals representing first cases in the family as a result of new, dominant mutations.
Although the genetic change causing NF2 is present at conception, the clinical manifestations occur over many years. The typical age of onset of symptoms is in the late teens to early 20s, but the age range covers the entire life span, to include congenital forms in infancy through the elderly. [11, 12, 13] Some evidence indicates that age of onset of clinical symptoms is lower in maternally transmitted NF2. While NF2 is quite variable in severity from person to person, family studies have shown some intrafamilial consistency in age of onset. Somatic mosaicism for the NF2 mutation in sporadic cases may also complicate the clinical picture, resulting in underdiagnosis or late diagnosis.
Prognosis
The prognosis of neurofibromatosis type 2 (NF2) depends on a number of factors, including age of symptom onset, degree of hearing deficit, and number and location of various tumors. Although age of onset is relatively similar within families, the age range can vary from 2-70 years. While the tumors themselves are relatively indolent and do not undergo malignant transformation, studies performed in the late 1980s and early 1990s showed clearly that significant rates of mortality and morbidity are associated with the diagnosis of NF2.
One such study suggested that the survival from the time of actual diagnosis averages 15 years [14] ; however, this may be evolving with improved diagnosis, surgical techniques, surveillance, screening, and recognition of mild disease (due in part to increased physician awareness and availability of molecular diagnostic options). [15] In a more recent study of 1,192 patients with NF2, increased mortality is associated with early age at diagnosis (< 20 years old) and with the presence of intracranial meningiomas. [15] Genetically, reduced mortality is associated with slice-site or missense mutations compared to patients with truncating mutations, and also reduced in mosaic patients compared to non-mosaic patients with NF2. [15]
Morbidity and mortality
Vestibular schwannomas are the most common and well-recognized feature of NF2 leading to significant morbidity. Symptoms of tinnitus, gradual hearing loss, and even vestibular dysfunction are frequently the initial signs of NF2. Although unilateral hearing loss is the most frequently presenting symptom, bilateral deafness would be expected to eventually occur in most affected individuals. Untreated vestibular schwannomas can extend locally and may result in brainstem compression, hydrocephalus, and, occasionally, facial nerve palsy.
Dumbbell-shaped spinal cord schwannomas are quite common in NF2 and result in significant morbidity; they present a great therapeutic challenge. Spinal cord ependymomas, astrocytomas, and meningiomas also occur, but less frequently. Intracranial meningiomas, on the other hand, are a frequent finding; they may be asymptomatic, or they may cause a variety of symptoms and CNS deficits. [16]
Nonvestibular schwannomas occur in more than half of patients and are often diagnosed in patients with an earlier age at diagnosis of NF2. Cranial nerves III and V are most commonly involved, but the rare occurrence of jugular foramen schwannomas potentially impacting the glossopharyngeal, vagus, and/or spinal accessory nerves may lead to dysphagia, esophageal dysmotility, hoarseness, or aspiration.
On the other hand, nonvestibular schwannomas in patients with NF2 tend to be more indolent and to grow slowly over time. This can complicate treatment decision making, since options include surgery, radiation therapy, and watchful waiting. [17]
Posterior subcapsular, or juvenile, cataracts can predate CNS symptomatology. These cataracts may progress over time, leading to decreased visual acuity. A fair percentage of affected individuals are found to have retinal hamartomas or epiretinal membranes that may or may not be visually significant.
Sensory motor polyneuropathy is seen in some individuals with NF2 who may or may not have identifiable tumors along the length of the peripheral nerve(s) of interest. Early peripheral nerve abnormalities, such as dorsal root ganglia hypertrophy, may be seen in young children early in the course of NF2, but there are likely additional factors that contribute to the development of neuropathy later in the clinical course. [18]
Patient Education
Patients and at-risk family members should be made aware of specific symptoms, such as tinnitus, hearing deficits, focal weakness, sensory changes, or balance problems, that might suggest tumor growth and should prompt immediate medical attention.
Patients with vestibular schwannomas should be cautioned about diving and underwater activities, because of increased risks for disorientation and potential for drowning.
Patients and their families may be referred to neurofibromatosis (NF)-specific regional and national support groups for continuous updates on advances in treatment, as well as for emotional support. Neurofibromatosis Network (nfnetwork.org), for example, can be reached at the toll-free number 1-800-942-6825.
The Children's Tumor Foundation (www.ctf.org) has a toll-free number (1-800-323-7938) for information and to sign up for their newsletter.
Other online resources include the NIH Web site and an NF2 person-to-person support group known as the NF2 crew (www.nf2.crew.org).
-
Subcutaneous and cutaneous lesions in a young man with neurofibromatosis type 2; note paucity of cafe-au-lait spots.
-
Right neck mass in a patient with neurofibromatosis type 2.
-
Facial asymmetry, OS proptosis, and exotropia, as well as several subcutaneous lesions on the forehead and face, in a 20-year-old man with neurofibromatosis type 2.
-
Posterior cervical scar from cord lesion resection, thoracic scoliosis, and subcutaneous masses in a young adult with neurofibromatosis type 2.
-
Meningioma to the left of midline in a patient with neurofibromatosis type 2.
-
Multiple meningiomas (on the left) on the surface of the brain in a patient with neurofibromatosis type 2.
-
Bilateral acoustic neuromas in a patient with neurofibromatosis type 2.
-
Bilateral acoustic neuromas and a left-sided meningioma in a patient with neurofibromatosis type 2.
-
Small ependymoma in a patient with neurofibromatosis type 2.
-
Multiple meningiomas in a patient with neurofibromatosis type 2.









