Background
Xeroderma pigmentosum (XP) is a group of rare autosomal-recessive inherited disorders characterized by extreme skin sensitivity to ultraviolet (UV) light, abnormal skin pigmentation, and high frequency of skin cancers, especially on sun-exposed skin (see image below). Dermatologic changes are the most conspicuous findings and are mandatory for the diagnosis. [1]
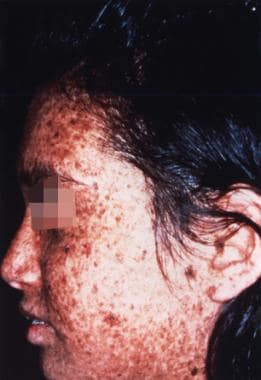 Sunlight-induced dermatologic abnormalities in a patient with xeroderma pigmentosum.
Sunlight-induced dermatologic abnormalities in a patient with xeroderma pigmentosum.
Neurologic involvement is often part of a phenotypic spectrum. Albert Neisser first described neurologic abnormalities associated with xeroderma pigmentosum in 1883. Neurologic involvement in xeroderma pigmentosum was emphasized in 1932 by De Sanctis and Cacchione, who described 3 brothers with xeroderma pigmentosum who had intellectual disability, dwarfism, and gonadal hypoplasia. Subsequently, some authors referred to xeroderma pigmentosum associated with CNS abnormalities as De Sanctis-Cacchione syndrome.
In 1987, Kraemer et al reviewed clinical characteristics of 830 patients with xeroderma pigmentosum who were described in 297 articles. These researchers found neurologic abnormalities in 152 (18%) of these patients, a rate similar to those in other reports. Among patients with nervous system involvement, the most common abnormality was intellectual disability (80% of subjects with neurologic involvement). Among these patients, the median intelligence quotient (IQ) score was 45, and the range was 15-81. The second most common neurologic abnormality was spasticity or ataxia (30% of subjects with neurologic involvement), followed by microcephaly (24%). [2]
Analysis of 106 patients with xeroderma pigmentosum followed at the National Institutes of Health identified a progressive neurologic degeneration in 25 (24%) patients, and 16 of these patients had complementary group XP-D. [3]
Pathophysiology
Cleaver's seminal work in 1968 elucidated the pathophysiology of xeroderma pigmentosum by demonstrating defective DNA repair. Further studies of this defect led to significant progress in the understanding of nucleotide excision repair (NER) mechanisms under normal and pathologic conditions. [4]
UV radiation induces cross-linking (dimerization) between thymine nucleotides. After exposure to UV light, normal cultured cells identify and excise the UV-induced thymine dimers and insert undamaged nucleotides after DNA synthesis and ligation. This repair process, known as unscheduled DNA synthesis, is deficient in xeroderma pigmentosum. Cell complementation analysis of cultured cells from patients with xeroderma pigmentosum demonstrated that xeroderma pigmentosum was genetically heterogeneous for the ability to repair UV-induced thymine dimers.
Fibroblasts from different patients with xeroderma pigmentosum were fused, and DNA repair after UV exposure was assayed. Correction of DNA repair deficiency in the fused cells indicates that each cell line has a unique abnormality of DNA repair. This finding led to identification of 7 specific complementation groups (A through G).
The genes that are responsible for defective NER in each xeroderma pigmentosum complementary group are highly conserved; homologous genes have been discovered in several species ranging from yeast to mammals.
Two overlapping pathways for NER have been proposed: the rapid transcription-coupled repair directed at the transcribed strand and slower global genome repair, which also includes the nontranscribed strand. [5] Most xeroderma pigmentosum complementary groups are defective in both pathways. The complementary group C (XP-C) is a notable exception in which only global genome repair is defective.
The xeroderma pigmentosum variant complementation group (XP-V) has normal unscheduled DNA synthesis after UV exposure. However, the ability to repair DNA is reduced after adding caffeine to cultured cells. This defect is caused by mutations in the (pol)eta polymerase, which initiates translesion synthesis of UV-damaged DNA in an error-free manner.
Xeroderma pigmentosum is a multisystem disorder; sun-exposed skin and eyes (ie, eyelids, conjunctivae) are the most affected tissues. Cutaneous photosensitivity and early development of skin cancer is caused by defective DNA repair. [6]
CNS involvement is due to premature neuronal death.
Necrosis in tissues that are not exposed to UV light suggests that these cells in patients with xeroderma pigmentosum are unable to repair DNA damage from other mutagens (eg, reactive oxygen species, other free radicals). Neurodegeneration probably results from accumulating mutations due to cells' inability to repair DNA damage. Increased oxidative damage in neurons due to abnormal function of free radical scavengers, such as superoxide dismutase, has been suggested.
The presence of neurologic abnormalities correlates with the degree of NER repair defect; patients with the greatest impairment of DNA repair are more prone to develop neurodegeneration.
Pathologic studies showed diffuse neuronal loss without other histologic hallmarks. Selective degeneration of dopaminergic neurons has been reported in some patients who were affected neurologically. Diffuse axonal loss was seen in the peripheral nerves in patients with clinical evidence of polyneuropathy.
Epidemiology
Frequency
XP-C and XP-D are the most common complementary forms, representing 30% and 20% of all xeroderma pigmentosum cases, respectively. XP-A is rare.
The worldwide frequency of xeroderma pigmentosum is estimated at 1 case in 250,000 population. Frequencies of complementary groups vary significantly in different populations. XP-A accounts for as many as 40% of all cases in Japan. Other complementary groups, with the exception of XP-V (in which all patients have only dermatologic manifestations), are rare. For example, only 3 cases of the XP-B type have been reported.
Mortality/morbidity
Skin cancer represents the major morbidity in xeroderma pigmentosum.
-
The median age of the first cutaneous cancer in xeroderma pigmentosum (most commonly basal cell or squamous cell carcinoma) is 8 years. In striking contrast, the mean age for squamous cell carcinoma in the general population is 58 years.
-
The incidence of malignant melanoma in patients with xeroderma pigmentosum who are younger than 20 years is 2000-fold higher than in an age-matched US population.
-
Skin tumors are typically multiple, and patients with as many as 100 tumors have been reported. This may result in disfigurement in severely affected subjects.
Keratitis, together with squamous cell tumors of the conjunctiva and corneoconjunctival junction, is a major source of ophthalmologic morbidity. [7]
Other malignancies also occur at increased frequency in patients with xeroderma pigmentosum. The frequency of inner organ neoplasms, including malignant brain tumors, is estimated to be increased 20-fold compared to subjects without xeroderma pigmentosum.
Intellectual disability (or dementia in subjects with adult-onset neurologic deterioration), hearing loss, spasticity, ataxia, and polyneuropathy are the most common morbidity factors in the subset of patients who have with neurologic impairment.
-
As many as 50% of patients with XP-D manifest neurologic deterioration. Neurologic involvement is rare in patients with XP-C, the most common complementary group in the United States.
-
Seizures are common and epilepsy may be present in almost 25% of all patients.
-
Neurologic symptoms are progressive and may result in severe disability.
Many patients become bedridden and incontinent. Some have significant cachexia in the terminal stages despite adequate caloric intake.
Urinary tract infection, sepsis, and aspiration pneumonia are potential complications.
Patients with early onset of neurologic symptoms tend to have more profoundly defective DNA repair, making them more susceptible to skin and inner organ tumors. The median age at death in xeroderma pigmentosum patients with signs of neurodegeneration was 29 years and in patients without any additional neurologic abnormalities was 38 years. [3]
Kraemer et al constructed the Kaplan-Meier survival curve for patients with xeroderma pigmentosum. The following were estimated:
-
90% probability of surviving to age 13 years
-
80% probability of surviving to 28 years
-
70% probability surviving to 40 years
-
Overall, life expectancy of patients with xeroderma pigmentosum reduced by 30 years
Various comorbid cancers usually cause death.
The most common cause of death in xeroderma pigmentosa patients is skin cancer, at 34%, followed by cancer of internal organs, at 17%.
Progressive neurodegeneration can be a direct cause of death in approximately one third of patients.
Race-, sex-, and age-related demographics
All ethnic groups are affected similarly.
Both sexes are affected equally.
Only 5% of patients manifest the first symptoms after age 14 years.
The median age of symptom onset is approximately 2 years.
Prognosis
Xeroderma pigmentosum is a progressive condition with a high incidence of cutaneous and inner organ tumors.
Progressive neurologic deterioration may occur.
The average life expectancy is reduced by 30 years, and neoplasms are usually the cause of death.
Patient Education
Patients and caregivers should be educated concerning the nature of the disease, the photosensitivity, the high incidence of cancer, and the necessity to avoid UV exposure.
Discuss skin protection (see Activity).
-
Sunlight-induced dermatologic abnormalities in a patient with xeroderma pigmentosum.
-
Typical skin manifestation of xeroderma pigmentosum with numerous areas of hypopigmentation and freckles (ie, solar lentigines) with different intensities of pigmentation.
-
Axial T2-weighted MRI of the brain of a 47-year-old woman with xeroderma pigmentosum, complementary group D. She developed progressive ataxia and dementia at age 44 years. Note severe atrophy and normal signal from the white matter.
-
Coronal T1-weighted MRI image of the brain of a 47-year-old woman who developed progressive ataxia and dementia at age 44 and was found to have xeroderma pigmentosum, complementary group D (see image above). Severe diffuse atrophy involves the brain stem and cerebellum.






