Background
Dystonia is a syndrome of sustained muscle contractions of agonist and antagonist muscles, usually resulting in twisting, torsional, and repetitive movements or abnormal postures. [1] It can either be primary or secondary. Primary torsion dystonia (PTD) is dystonia in isolation without brain degeneration and without an acquired cause. Secondary dystonia includes a heterogenous group of etiologies including inherited (with and without brain degeneration) and acquired neurologic disorders. The phenotypic spectrum associated with PTD is broad, from early-onset generalized to adult-onset focal dystonia. [3, 4]
The first description of what is now considered primary, or idiopathic, torsion dystonia was described by Schwalbe in 1908. In 1911, Oppenheim termed this same condition dystonia musculorum deformans (DMD) or dysbasia lordotica progressiva. [2] Initially believed to be a manifestation of hysteria, idiopathic torsion dystonia is now established as a specific neurologic entity with a well-established genetic basis. DMD and Oppenheim disease are terms now used for childhood- and adolescent-onset dystonia due to the DYT1 gene.
Classification of dystonia
At the present time there are 25 types of genetically determined dystonias. Several classification schemes have been used to categorize the various forms of dystonia. One common scheme is based on genetic features, including mode of inheritance and molecular genetic data. There is also a topographic classification where torsion dystonia may be described as focal, segmental, multifocal, or generalized, depending on which anatomic distribution of the symptoms (see Table 1).
Table 1. Anatomic Distribution of Primary Torsion Dystonia (Open Table in a new window)
Focal |
Body Site |
Segmental |
two or more contiguous body regions |
Multifocal |
two or more noncontiguous body regions |
Generalized |
involving atleast one leg, the trunk and another body region |
Hemidystonia |
involving one side of the body |
In addition, depending on the clinical features, dystonias can be divided into two main groups: isolated dystonia or combined dystonia. Combined dystonia can be classified into three sub-types: those accompanied by parkinsonism, by myoclonus, or by a mixed pattern of various hyperkinetic movements.
Genetically defined isolated dystonias include TOR1A/DYT1, TUBB4/DYT4, THAP1/DYT6, PRKRA/DYT16, CIZ1/DYT23, ANO3/DYT24, and GNAL/DYT25. Combined dystonias, accompanied by parkinsonism with known genetic loci include TAF1/DYT3, GCH1/DYT5a, TH/DYT5b, and ATP1A3/DYT12. Genetically determined dystonias that are accompanied by myoclonus include SGCE/DYT11, whereas dystonias that accompany a mixed pattern of hyperkinetic disorders include MR-1/DYT8, PRRT2/DYT10, and SLC2A1/ DYT18. [51]
Sometimes, combined dystonias are also classified depending on whether the symptoms are continually and continuously present or whether they are paroxysmal. Generic forms of common, persistent combined dystonias are GCH1/ DYT5a, TH/DYT5b, SGCE/DYT11, APT1A3/DYT12, and TAF1/ DYT3. Genetically defined paroxysmal combined dystonias include MR-1/DYT8, PRRT2/DYT10, and SLC2A1/DYT18. [51]
DYT1 (early-onset generalized dystonia)
DYT1 are caused by a 3-base pair in-frame deletion within the coding region of the TOR1A (torsinA) gene located on chromosome 9q34. TorsinA is expressed at high levels in neuronal cytoplasm of specific neuronal populations in the adult human brain, including the SN, thalamus, cerebellum, hippocampus, and neostriatum.
DYT1 is the most common hereditary dystonia. Phenomenologically, it is an isolated dystonia. Some degree of genetic anticipation in regards to the age of onset and disease severity has been noted in DYT1. It is especially common among the Ashkenazi Jewish population.
In most instances, DYT1 symptoms often start with a focal dystonia as talipes equinovarus of one leg in early childhood, typically around 6 years of age. The dystonic posturing then gradually progresses with age to other extremities and trunk muscles by the early teens. There is obvious asymmetry to the dystonia, with involvement of the extremities on the dominant side along with the ipsilateral sternocleidomastoid muscle. In these patients, interlimb coordination and locomotive movements are not affected at all. Moreover, intellectual, mental, and psychological functions are completely intact in these patients.
Based on clinical characteristics, DYT1 can be classified into two types: the postural type with appendicular and truncal dystonias, or the action type, which is associated with violent dyskinetic movements in addition to dystonic posture.
DYT5 (dopa-responsive dystonia)
Hereditary progressive dystonia with marked diurnal fluctuation, or Segawa disease, is an autosomal dominantly inherited dopa-responsive dystonia (DRD) caused by heterozygous mutations of the GCH1 gene located on chromosome 14q22.1-q22.2. DYT5 shows a marked female predominance in the young. In contrast, adult-onset cases show a male predominance
Onset is around 6 years of age, mostly with rigid talipes equinovarus of one foot not dissimilar to DYT1. With age, it expands to other limbs and trunk muscles by the midteens with progressive rigidity. Starting around age 10 years, postural tremor of 8 to 10 Hz appears. These symptoms show marked diurnal fluctuations, worsening through the day and almost absent in the early morning. However, this fluctuation decreases with age in the late teens, and is no longer apparent in early adulthood, when symptoms become static. Clinically, DYT5 is also classified into two types: postural and action. Patients with the action type develop dystonic movements of one extremity or the neck (action retrocollis) in addition to dystonic, and show focal or segmental dystonia during the teenage years.
Pathophysiology
In DYT1 and the other genetic dystonias, no consistent histologic or biochemical abnormalities have been identified. However, perinuclear inclusion bodies have been described in the midbrain reticular formation and in the periaqueductal gray matter in 4 patients in whom DYT1 was clinically documented and genetically confirmed. [5] This is in contrast, however, to the secondary forms of dystonia that are frequently associated with macroscopic structural lesions of the basal ganglia and thalamus.
As such, in primary torsion dystonias and other genetic dystonias no discernible abnormalities are seen on structural neuroimaging studies. Abnormal brain networks have been described in different functional imaging studies; substantial evidence implicates dysfunction in dopaminergic pathways in the pathophysiology of primary torsion dystonia. [6, 7]
Besides motor control difficulties, defective sensory processing and sensory abnormalities are described, [8, 9] but these findings are inconsistent.
While the exact pathogenesis of dystonia is unknown, based on the current models of basal ganglia circuitry, some form of electro-biochemical dysfunction at the basal ganglia level has been proposed as the underlying unifying mechanism behind various forms of dystonia. [10] Such dysfunctions may involve direct and indirect pathways and result in impaired center-surround inhibition at the cortical level.
See the image below for a diagram of the basal ganglia circuitry dysfunction in dystonia.
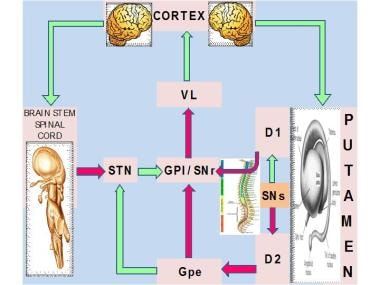 Idiopathic torsion dystonia. Major nuclear complex of the basal ganglia is the striatum, which is composed of the caudate and putamen. The striatum receives glutamatergic input from the cerebral cortex and dopaminergic input from the substantia nigra pars compacta (SNc). Two types of spiny projection neurons receive cortical and nigral inputs: those that project directly and those that project indirectly to the internal segment of the globus pallidus (GPI), which is the major output site of the basal ganglia. Complementary action of both of these pathways regulates the overall function of the GPI. The GPI, which, in turn, provides tonic inhibitory (ie, gamma-aminobutyric acid [GABA]–ergic) discharges downstream into the thalamic nuclei that project to the frontal cortical and other CNS areas. Direct pathway (D1) inhibits the substantia nigra pars reticulata (SNr) and the GPI, which are the major output sites, resulting in a net disinhibition and facilitation of thalamocortical circuits. Indirect pathway (D2), through serial connections with the globus pallidus pars externa (GPe) and the subthalamic nucleus (STN), is excitatory to the GPI, resulting in further inhibitory action on thalamocortical pathways. In this model, the mean discharge rate of the GPI is the key factor that determines a hypokinetic or hyperkinetic movement disorder. Increased inhibitory influences of the GPI on the thalamocortical circuitry result in hypokinetic disorders, such as Parkinson disease, whereas decreased GPI activity results in hyperkinetic disorders, such as hemiballismus. VL = ventrolateral thalamus.
Idiopathic torsion dystonia. Major nuclear complex of the basal ganglia is the striatum, which is composed of the caudate and putamen. The striatum receives glutamatergic input from the cerebral cortex and dopaminergic input from the substantia nigra pars compacta (SNc). Two types of spiny projection neurons receive cortical and nigral inputs: those that project directly and those that project indirectly to the internal segment of the globus pallidus (GPI), which is the major output site of the basal ganglia. Complementary action of both of these pathways regulates the overall function of the GPI. The GPI, which, in turn, provides tonic inhibitory (ie, gamma-aminobutyric acid [GABA]–ergic) discharges downstream into the thalamic nuclei that project to the frontal cortical and other CNS areas. Direct pathway (D1) inhibits the substantia nigra pars reticulata (SNr) and the GPI, which are the major output sites, resulting in a net disinhibition and facilitation of thalamocortical circuits. Indirect pathway (D2), through serial connections with the globus pallidus pars externa (GPe) and the subthalamic nucleus (STN), is excitatory to the GPI, resulting in further inhibitory action on thalamocortical pathways. In this model, the mean discharge rate of the GPI is the key factor that determines a hypokinetic or hyperkinetic movement disorder. Increased inhibitory influences of the GPI on the thalamocortical circuitry result in hypokinetic disorders, such as Parkinson disease, whereas decreased GPI activity results in hyperkinetic disorders, such as hemiballismus. VL = ventrolateral thalamus.
Epidemiology
Frequency
The exact relative frequencies of primary and secondary forms of dystonia remain unknown.
The prevalence of primary torsion dystonia is difficult to estimate because of the variation in its phenotypic expression and the tendency for mild cases to go undiagnosed. In Rochester, Minnesota, the prevalence was calculated to be approximately 34 per million persons for generalized dystonia and 295 per million persons for all focal dystonia from a study conducted in 1980s. [11] Late-onset focal primary dystonia was 10 times more common than early-onset generalized primary torsion dystonia. [11]
Several large studies have shown that early-onset primary torsion dystonia is 5-10 times more common in Ashkenazi Jews than in people who were not Jewish or in Jewish individuals not of Ashkenazi heritage. Subsequent studies have found a wide range in the prevalence of dystonia from 6-7,320 persons per million population. [12, 13]
In a European collaborative study (the Epidemiological Study of Dystonia in Europe [ESDE]), investigators found a crude annual prevalence of 15.2 cases per 100,000 individuals, the majority of whom had focal dystonia at a rate of 11.7 cases per 100,000 individuals. [14]
Race
Childhood- and adolescent-onset primary dystonia is more common in Jews of Eastern European or Ashkenazi ancestry than in other groups and seemingly rare in Far Eastern populations.
-
Many cases of early primary torsion dystonia, especially those among non-Jewish populations, are not due to the TOR1A GAG deletion in DYT1. The DYT6 locus was identified by means of linkage analysis in 15 affected members from 2 Swiss Mennonite families. [15]
-
A genome-wide search for primary torsion dystonia in a large family from central Italy in whom 11 members were definitely affected revealed a novel locus, namely, DYT13. [16]
Gender
In a large study of 957 cases of primary dystonia from Europe, segmental and focal dystonias had notable female predilections. This finding suggested that patients with focal dystonia should not be treated as a homogeneous group and that sex-linked factors may play a role. [14]
-
Idiopathic torsion dystonia. Major nuclear complex of the basal ganglia is the striatum, which is composed of the caudate and putamen. The striatum receives glutamatergic input from the cerebral cortex and dopaminergic input from the substantia nigra pars compacta (SNc). Two types of spiny projection neurons receive cortical and nigral inputs: those that project directly and those that project indirectly to the internal segment of the globus pallidus (GPI), which is the major output site of the basal ganglia. Complementary action of both of these pathways regulates the overall function of the GPI. The GPI, which, in turn, provides tonic inhibitory (ie, gamma-aminobutyric acid [GABA]–ergic) discharges downstream into the thalamic nuclei that project to the frontal cortical and other CNS areas. Direct pathway (D1) inhibits the substantia nigra pars reticulata (SNr) and the GPI, which are the major output sites, resulting in a net disinhibition and facilitation of thalamocortical circuits. Indirect pathway (D2), through serial connections with the globus pallidus pars externa (GPe) and the subthalamic nucleus (STN), is excitatory to the GPI, resulting in further inhibitory action on thalamocortical pathways. In this model, the mean discharge rate of the GPI is the key factor that determines a hypokinetic or hyperkinetic movement disorder. Increased inhibitory influences of the GPI on the thalamocortical circuitry result in hypokinetic disorders, such as Parkinson disease, whereas decreased GPI activity results in hyperkinetic disorders, such as hemiballismus. VL = ventrolateral thalamus.
Tables
Focal |
Body Site |
Segmental |
two or more contiguous body regions |
Multifocal |
two or more noncontiguous body regions |
Generalized |
involving atleast one leg, the trunk and another body region |
Hemidystonia |
involving one side of the body |
| Type | Designation | Mode of Inheritance | Gene | Gene Locus | OMIM# |
|---|---|---|---|---|---|
DYT1 |
Early-onset generalized | Autosomal dominant | TOR1A | 9q.34.11 | 128100 |
| DYT2 | Early-onset generalized | Autosomal recessive | Uknown | Uknown | 224500 |
| DYT3 | X-linked dystonia parkinsonism (Lubag syndrome) | X-chromosomal recessive | TAF1 | Xq13.1 | 314250 |
| DYT4 | Torsion dystonia (Whispering dysphonia) | Autosomal dominant | TUBB4A | 19p13.3 | 128101 |
| DYT5a | Dopa-responsive dystonia (Segawa disease) | Autosomal dominant | GCH1 | 14q22.1–22.2 | 128230 |
| DYT5b | Dopa-responsive dystonia | Autosomal recessive | TH | 11p15.5 | 605407 |
| DYT6 | Adolescent-onset mixed phenotype | Autosomal dominant | THAP1 | 8p11.21 | 602629 |
| DYT7 | Paroxysmal dystonic choreoathetosis | Autosomal dominant | Unknown | 18p | 602124 |
| DYT8 | Paroxysmal kinesigenic, nonkinesigenic dyskinesia | Autosomal dominant | MR-1 | 2q33–35 | 118800 |
| DYT9 | Paroxysmal choreoathetosis with spasticity | Autosomal dominant | CSE | 1p | 601042 |
| DYT10 | Paroxysmal kinesigenic dystonia | Autosomal dominant | PRRT2 | 16q11.2–12.1 | 128200 |
| DYT11 | Myoclonus dystonia | Autosomal dominant | SGCE | 7q21.3 | 159900 |
| DYT11 | Myoclonus dystonia | Autosomal dominant | DRD2 | 11q23.2 | 159900 |
| DYT12 | Rapid-onset dystonia parkinsonism (syndrome) | Autosomal dominant | ATP1A3 | 19q12–13.2 | 128235 |
| DYT13 | Early- and late-onset focal or craniocervical dystonia | Autosomal dominant | Unknown | 1p36.32-p36.13 | 607671 |
| DYT14 | Dopa-responsive generalized dystonia | ||||
| DYT15 | Myoclonus-dystonia | Autosomal dominant | Unknown | 18p11 | 607488 |
| DYT16 | Dystonia-parkinsonism syndrome | Autosomal recessive | PRKRA | 2q31.2 | 612067 |
| DYT17 | Adolescent onset | Autosomal recessive | Unknown | 20p11.2-q13.12 | 612406 |
| DYT18 | Paroxysmal exertion-induced dyskinesia | Autosomal dominant | SLC2A1 | 1p34.2 | 612126 |
| DYT19 | Paroxysmal kinesigenic dyskinesia 2 | Autosomal dominant | Unknown | 16q13-q22.1 | 611031 |
| DYT20 | Paroxysmal nonkinesigenic dyskinesia 2 | Autosomal dominant | Unknown | 2q31 | 611147 |
| DYT21 | Late-onset torsion dystonia | Autosomal dominant | Unknown | 2q14.3-q21.3 | 614588 |
| DYT22 | Unknown | Unknown | Not listed | ||
| DYT23 | Adult-onset cervical dystonia | Autosomal dominant | CIZ1 | 9q34 | 614860 |
| DYT24 | Focal dystonia | Autosomal dominant | ANO3 | 11p14.2 | 615034 |
| DYT25 | Adult-onset focal dystonia | Autosomal dominant | GNAL | 18p11.21 | 615073 |



