Practice Essentials
Glycogen storage diseases (GSDs) are inherited disorders due to enzymatic defects that prevent breakdown of stored glycogen into glucose. GSD type I, also known as Von Gierke disease, is an autosomal recessive disorder, divided into two subtypes: type Ia and type Ib. GSD type Ib is caused by a mutation in the glucose-6-phosphate transporter gene (G6PT1, also known as SLC37A4), [1] leading to a defect in the last step of glycogenolysis, resulting in fat accumulation and dysfunction of the liver and kidneys, infantile hypoglycemia, hepatomegaly, and life-threatening seizures if not treated early. [2]
Signs and symptoms of type Ib glycogen storage disease
Typically, the diagnosis is made in infancy with the symptoms and signs noted early in life. GSD type Ia and Ib both have the hallmark feature of hypoglycemia, which can lead to life-threatening seizures and prompts the workup early in infancy. Other symptoms and signs include the following:
-
Hypotonia with poor metabolic control
-
Frequent skin and pulmonary infections
-
"Crohn's disease like"-inflammatory bowel disease [3]
-
Hepatomegaly
-
Nephromegaly
-
Delayed growth and development
-
Characteristic facies (doll-like face with fat cheeks)
-
Symptoms of inflammatory bowel disease, including cramps, fever, and abdominal pain
See Presentation for more detail.
Diagnosis of type Ib glycogen storage disease
The following studies are typically included in the workup:
-
Fasting glucose measurement
-
Lipid panel
-
Liver function test (elevated early in disease course, but typically normal in GSD I)
-
Complete blood cell count with differential (to detect neutropenia common in GSD type Ib)
-
Urine studies
-
Uric acid level (elevated)
-
Creatine kinase measurement (typically normal)
-
Electromyelography
-
Muscle biopsy
-
Ischemic forearm test
Genetic testing for SLC37A4 gene mutation confirms the diagnosis; it can be performed for carrier testing and prenatal diagnosis.
See Workup for more detail.
Management of type Ib glycogen storage disease
Although there is no cure, a diet that avoids fasting to maintain normal glucose level is the mainstay of life-long treatment. Diets should be restricted of galactose and fructose, and include frequent small servings of simple carbohydrates day and night.
See Treatment for more detail.
Background
Glycogen storage diseases (GSD) are a group of inherited autosomal recessive disorders caused by genetic mutations that lead to the inability to breakdown and metabolize glycogen into glucose. The resultant inability to breakdown glycogen results in excessive buildup of glycogen in organs responsible for gluconeogensis and glycogenolysis, most importantly the liver and kidneys. In most cases, the defect has systemic consequences; however, in others the defect is limited to specific organs. Most patients experience muscle symptoms, such as weakness and cramps, although certain GSDs manifest as specific syndromes, such as hypoglycemic seizures or cardiomegaly.
The image below illustrates the metabolic pathways for carbohydrates.
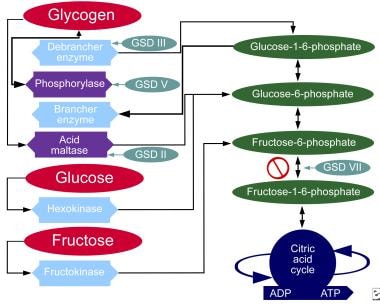 Metabolic pathways of carbohydrates.
Metabolic pathways of carbohydrates.
Although at least 14 unique GSDs are discussed in the literature, the 5 that cause clinically significant symptomology are Pompe disease (GSD type II, acid maltase deficiency), Cori disease (GSD type III, debranching enzyme deficiency), McArdle disease (GSD type V, myophosphorylase deficiency), Tarui disease (GSD type VII, phosphofructokinase deficiency), and Von Gierke disease (GSD type Ia, glucose-6-phosphatase deficiency or type Ib, glucose-6-phosphate transporter mutation). The remaining GSDs are not benign but are less clinically significant; therefore, the physician should consider the aforementioned GSDs when initially entertaining the diagnosis of a GSD. Interestingly, there also is a GSD type 0, which is due to defective glycogen synthase.
Von Gierke disease (GSD type Ia and Ib) was first reported in 1929 based on the autopsy findings in 2 children who had excessive hepatic and renal glycogen accumulation. Although 4 other cases were reported of excess glycogen storage in the livers of autopsy patients, it was not until 1978 when Narisawa et al were able to differentiate GSD type Ia and type Ib, recognizing type Ia was due to a deficiency in the G6Pase enzyme, and type Ib was due to deficiency in the G6P transporter. [4] GSD Ib, like other GSDs, is also an autosomal recessive disorder and causes significant end-organ disease with significant morbidity, and typically presents in infancy (by age 3-4 months).
Pathophysiology
GSD type Ib is an autosomal recessive condition caused by a mutation in the glucose-6-phosphate transporter gene (G6PT1, also known as SLC37A4). [1] Normally, in the terminal steps of both glyogenolysis and gluconeogensis, glucose-6-phosphate transporter (G6PT) permits movement of G6P from the cytoplasm into the lumen of the endoplasmic reticulum, presenting it to a G6Pase for breakdown to glucose and phosphate and release into the cytoplasm. Mutation of SLC37A4 results in deficiency of the G6P transporter (G6PT), which then prevents transport of G6P from the cytoplasm to the endoplasmic reticulum. This leads to hypoglycemia, glycogen accumulation, and dysfunction in the liver, kidneys, and intestinal mucosa. Of those individuals with GSD type I, 80% have genetic mutation of the G6Pase (type Ia) and 20% have mutation of the G6PT (type Ib).
Impaired gluconeogensis and glycogenolysis then leads to increased metabolites such as lactic acid, uric acid, lipids, and triglyceride. Like GSD type Ia, type Ib presents with hypoglycemia and lactic acidemia in infancy but more commonly at 3-6 months of age, along with growth retardation, hyperlipidemia, hyperuricemia, and lactic acidemia. Unlike type Ia, GSD type Ib results in neutropenia and myeloid dysfunction, causing recurrent bacterial infections. The underlying mechanisms for neutropenia and myeloid dysfunction are not completely understood. Patients with GSD-I (both type Ia and type Ib) do not generally have skeletal abnormalities or myopathy.
Epidemiology
International data
GSD I is a disorder with genetic mutations found in multiple ethnic groups and has an overall incidence of ~1/100,000. Among the Ashkenazi Jews, there is a relatively high prevalence (1/20,000). [5] Newer epidemiological studies found the Serbian population had an incidence of ~1/60,000 live births, which is the highest incidence of GSD Ib worldwide. [6]
Prognosis
Morbidity/mortality
Long-term outcomes have improved with appropriate dietary therapy, but due to ongoing complications, morbidity and mortality still exists and depends on age and duration of disease. Early in life, morbidity is due to hypoglycemia, which can lead to life-threatening seizures. With advancing age, hepatic and renal dysfunction due to glycogen accumulation may lead to hepatic and renal failure and hepatocellular adenoma. Individuals with GSD type Ib (unlikely GSD type Ia) also develop recurrent bacterial infections due to neutropenia and inflammatory bowel disease along with complications of liver adenomas. Other complications of GSD type Ib include the following:
-
Hepatic adenoma with potential malignant transformation
-
Recurrent pulmonary and skin infections
-
Secondary diabetes mellitus (may be a late complication)
-
Acute myelogenous leukemia (Pinsk et al suggest surveillance for acute myelogenous leukemia as a potential complication of GSD type Ib [7] )
-
Metabolic pathways of carbohydrates.




