Practice Essentials
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is the most common enzymatic disorder of red blood cells, affecting 400 million people worldwide. [1] Paul Carlson and colleagues first reported G6PD deficiency in 1956 while treating a patient previously identified as "primaquine sensitive." [2]
G6PD is an enzyme involved in the pentose monophosphate pathway. G6PD deficiency leads to free radical–mediated oxidative damage to red blood cells, which in turn causes hemolysis. [3] It is an X-linked recessive disorder, and thus more often affects males. G6PD deficiency has a high prevalence in people of African, Asian, and Mediterranean descent. The condition is polymorphic, with more than 400 variants.
Patients with G6PD-deficient alleles have a selective advantage against severe malaria; hence, G6PD deficiency is highly prevalent in populations where malaria is endemic.
Signs and symptoms of G6PD deficiency
Most patients with G6PD deficiency are asymptomatic. Neonatal jaundice may be seen in newborns.
Patients may experience episodes of intravascular hemolysis and consequent anemia, triggered by infections, medicines that induce oxidative stresses, fava beans, and ketoacidosis. Hemolysis begins 24-72 hours after exposure to oxidative stress. Patients with severe hemolysis present with weakness, tachycardia, jaundice, and hematuria.
The clinical presentation of G6PD deficiency includes a spectrum of hemolytic anemia ranging from mild to severe hemolysis in response to oxidative stress. The likelihood of developing hemolysis and its severity depend on the level of the enzyme deficiency, which in turn depends on the G6PD variant. [4, 5]
For patient education information, see the Newborn Jaundice Directory.
Workup in G6PD deficiency
Semi-quantitative tests
The fluorescent spot test is a direct test that measures the generation of reduced nicotinamide adenine dinucleotide phosphate (NADPH) from nicotinamide adenine dinucleotide phosphate (NADP+); the test is positive if the blood spot fails to show fluorescence under ultraviolet light. It is rapid, simple, sensitive, and inexpensive. [6, 7, 8]
The methemoglobin reduction test is a rapid indirect test that measures the reduced methemoglobin levels produced after NADPH oxidation. [6]
The cytofluorimetric test is a cytochemical typing assay that provides a fluorometric readout of the classic methemoglobin reduction test at the level of an individual red blood cell. [7]
Quantitative tests
Quantitative tests for G6PD activity are considered the criterion standard. The rate of NADPH generation is spectrophotometrically measured at a wavelength of 340 nm. The G6PD activity is finally expressed as G6PD IU/red blood cell and G6PD IU/hemoglobin ratios. [6, 7, 8]
Spectrophotometric quantitation may fail to detect deficiency in heterozygous females, due to residual activity in G6PD-sufficient cells. Regarding the identification of G6PD-deficient, as well as G6PD-sufficient, cells by a cytochemical method or cytofluorometry, these analyses are more sensitive in testing for G6PD deficiency in females. [9]
Management in neonates
Infants with prolonged neonatal jaundice as a result of G6PD deficiency should receive phototherapy. Exchange transfusion may be necessary in cases of severe neonatal jaundice.
Systematic assessment for the risk of severe hyperbilirubinemia should be performed before discharge in neonates in whom G6PD deficiency is suspected, to provide early and focused follow-up to prevent kernicterus (bilirubin encephalopathy). [10, 11, 12] Consultations with a hematologist are ideal for long-term follow up.
See Glucose-6-Phosphate Dehydrogenase (G6PD) Deficiency for more information on management in adults.
Pathophysiology
The G6PD enzyme is part of the pentose monophosphate shunt. It catalyzes the oxidation of glucose-6-phosphate and the reduction of nicotinamide adenine dinucleotide phosphate (NADP+) to nicotinamide adenine dinucleotide phosphate (NADPH). NADPH maintains glutathione in its reduced form, with glutathione acting as a scavenger for dangerous oxidative metabolites.
The pentose monophosphate shunt is the only source for NADPH in red blood cells. Therefore, red blood cells depend on G6PD activity to generate NADPH for protection. Consequently, red blood cells are more susceptible to oxidative stresses than other cells. In persons with G6PD deficiency, oxidative stresses can denature hemoglobin and cause intravascular hemolysis. Denatured hemoglobin can be visualized as Heinz bodies in peripheral blood smears processed with supravital staining. Heinz bodies are shown in the figure below.
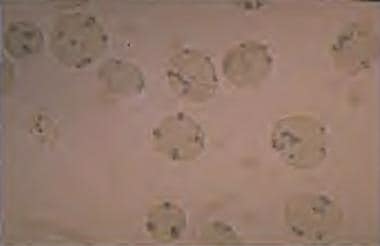 G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.
G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.
Drugs, chemical agents, infections, ingestion of fava beans, and ketoacidosis can trigger oxidative stress leading to hemolysis.
Jaundice in G6PD-deficient neonates is considered to be due to an imbalance between the production and conjugation of bilirubin, with a tendency toward inefficient bilirubin conjugation. Premature infants are at special risk for the bilirubin production-conjugation imbalance.
Etiology
G6PD deficiency is an X-linked recessive enzymopathy caused by a missense mutation in the housekeeping G6PD gene. [13] The pattern of inheritance is similar to that for hemophilia and color blindness; ie, males usually manifest the abnormality, and females are carriers. Females can be symptomatic if they are homozygous or if their normal X chromosome is inactivated.
The G6PD gene is located in the distal long arm of the X chromosome at the Xq28 locus. There have been 186 mutations documented in the G6PD gene. Most are single-base changes that result in an amino acid substitution. [3] These substitutions affect enzyme activity by decreasing intracellular stability of the protein or by affecting their catalytic activity. [14, 15, 16]
A large deletion in the G6PD gene is incompatible with life. Although small deletion mutation is rare, it has been reported and presents with severe G6PD deficiency. [16]
Specific G6PD alleles are associated with G6PD variants with different enzyme levels and, thus, different degrees of clinical disease severity. The variation in G6PD levels accounts for differences in sensitivity to oxidants.
The most common G6PD variants include G6PD A-, G6PD Mediterranean, G6PD Canton, and G6PD Union. [16]
G6PD A- occurs in high frequency in Africa, southern Europe, and North and South America. It is associated with lower enzyme levels and acute intermittent hemolysis. [4, 16, 17, 18]
G6PD Mediterranean is seen mainly in the Middle East, including Israel, and it accounts for almost all G6PD deficiency in Kurdish Jews. [4, 16, 19, 20, 21, 22, 23, 24, 25, 17, 18] It is characterized by enzyme deficiency that is more severe than that associated with G6PD A- alleles. Hemolysis after ingestion of fava beans (favism) is most frequently associated with the Mediterranean variant of G6PD deficiency.
G6PD Canton is seen mainly in China, and G6PD Union is seen worldwide.
G6PD B is the wild type of allele (normal variant). The G6PD A+ variant is associated with high enzyme levels and, hence, no hemolysis.
Severe forms of G6PD deficiency are associated with chronic nonspherocytic hemolytic anemia. Mutations causing this anemia commonly cluster in exon 10, a region important for protein dimerization. [16, 26]
The World Health Organization has classified the different G6PD variants according to the degree of enzyme deficiency and severity of hemolysis, as follows: [27]
-
Class I - Severe enzyme deficiency, chronic nonspherocytic hemolysis
-
Class II - Severe enzyme deficiency (1-10% residual activity), intermittent acute hemolysis
-
Class III - Moderate enzyme deficiency (10-60% residual activity), intermittent acute hemolysis
-
Class IV - No enzyme deficiency (60-150% activity)
-
Class V - Increased enzyme activity (>150%)
Epidemiology
G6PD deficiency is prevalent worldwide. In the United States, African Americans are primarily affected, with a prevalence of about 10%; however it is also seen among Italians (especially those of Sardinian ancestry), Greeks, Turkish people, Southeast Asians, people of Asian ancestry, and Sephardic Jews. [12]
Internationally, the geographic prevalence of the disorder correlates with the distribution of malaria. The highest prevalence rates (with gene frequencies from 5-25%) are found in the following regions:
-
Tropical Africa
-
The Middle East
-
Tropical and subtropical Asia
-
Some areas of the Mediterranean
-
Papua New Guinea
The heterogeneity of polymorphic G6PD variants is proof of their independent origin, and it supports the notion that they have been selected by a common environmental agent, in keeping with the concept of convergent evolution.
G6PD deficiency affects all races, although the severity of G6PD deficiency varies significantly among racial groups. The highest prevalence is among the people of African, Asian, or Mediterranean descent. Variants producing severe deficiency primarily occur in the Mediterranean population. African populations have milder hemolysis due to higher enzyme levels.
Prevalences were addressed in a study by Koromina et al, which also indicated that the frequency of genetically encoded G6PD deficiency is greatest among Africans; in the investigators’ estimate, the prevalence is 12.4% in African males and 3.3-5.9% in females. The prevalence is also considered high in Asia, being 4.4% in South Asian males and 0.5-1.2% in East Asian females. In contrast, the prevalence in Finnish females is 0.02-0.04%, and in the Amish population (males and females), 0%. [28]
G6PD deficiency is an X-linked inherited disease that primarily affects men. Women may be affected if they are homozygous, which occurs in populations in which the frequency of G6PD deficiency is quite high. Heterozygous women (carriers) can experience clinical disease as a result of X-chromosome inactivation, gene mosaicism, or hemizygosity.
Prognosis
Many people with G6PD deficiency are asymptomatic. However, case reports of acute massive hemolysis with jaundice have been reported, especially in the neonatal period, leading to kernicterus and fatalities. [29, 30, 31, 26, 32]
Kernicterus, or bilirubin encephalopathy, is a rare complication of neonatal jaundice complicated by G6PD deficiency. Kernicterus, although infrequent, has about a 10% mortality rate and 70% long-term morbidity rate, usually evident in infants with a bilirubin level higher than 20 mg/dL. [10]
Massive hemolysis complicating G6PD deficiency has been reported in patients with hepatitis infections, specifically hepatitis A and E in the Indian subcontinent. [33]
A literature review by Lai et al suggested that G6PD deficiency is a risk factor for diabetes, with the risk being greater in men than in women (odds ratio of 2.22 vs 1.87, respectively). [34]
A study by Rostami-Far et al indicated that G6PD deficiency increases the likelihood of neonatal sepsis. The study involved 76 neonates with sepsis and 1214 without sepsis, with the prevalence of G6PD deficiency being significantly greater in the sepsis group than in the controls. [35]
Patient Education
The X-linked pattern of inheritance of G6PD deficiency and its clinical severity should be discussed with parents, and counseling with regard to their risk for having other children with the condition should be provided, especially in populations in which G6PD deficiency is highly prevalent. [11]
If a mother is heterozygous, the chance of recurrence is 50% with every subsequent male pregnancy. [14]
Parental-child G6PD deficiency self-care discussions are associated with better child health, and parental involvement in these discussions is facilitated by the thoroughness and clarity of patient education received from the provider. [11]
Additional resources are available at G6PD Deficiency Association - Favism.
-
G6PD deficiency: Heinz bodies in a peripheral smear stained with a supravital stain. Heinz bodies are denatured hemoglobin, which occurs in G6PD deficiencies and in unstable hemoglobin disorders.





