Practice Essentials
Chronic lymphocytic leukemia (CLL) is a monoclonal disorder characterized by a progressive proliferation and accumulation of mature yet functionally incompetent lymphocytes. The histologic sample in the image below portrays the appearance of these lymphocytes. CLL is the most common form of leukemia found in adults in Western countries. [1] Some patients die within several years of diagnosis, usually due to complications from CLL, but most patients survive for at least five years. [2]
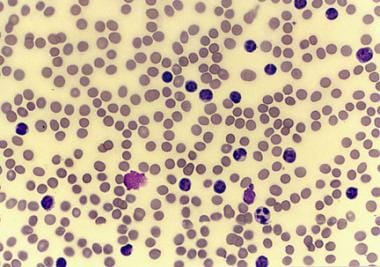 Peripheral smear from a patient with chronic lymphocytic leukemia, small lymphocytic variety.
Peripheral smear from a patient with chronic lymphocytic leukemia, small lymphocytic variety.
Signs and symptoms
Patients with CLL present with a wide range of signs and symptoms. The onset is insidious, and it is not unusual for CLL to be discovered incidentally when a blood cell count is performed for another reason; 25-50% of patients will be asymptomatic at time of presentation.
Signs and symptoms include the following:
-
Enlarged lymph nodes, liver, or spleen
-
Recurrent infections
-
Loss of appetite or early satiety
-
Abnormal bruising (late-stage sign)
-
Fatigue
-
Night sweats
See Presentation for more detail.
Diagnosis
Patients with CLL typically have a higher-than-normal white blood cell count, which is determined by a complete blood count (CBC). The most recent guidelines from the International Workshop for CLL (iwCLL) state that the diagnosis requires the presence of at least 5000 B-lymphocytes/μL for at least 3 months. [3] The clonality of the B-lymphocytes must be confirmed with flow cytometry. Other tests that may be helpful for diagnosis include bone marrow biopsy and ultrasonography of the liver and spleen. Immunoglobulin testing may be indicated for patients who develop repeated infections.
Staging
Two staging systems are used for CLL: Rai and Binet.
The Rai staging system categorizes patients into low-, intermediate-, and high-risk groups, as follows:
-
Low risk (formerly stage 0) – Lymphocytosis in the blood and marrow only (25% of presenting population) [4]
-
Intermediate risk (formerly stages I and II) – Lymphocytosis with enlarged nodes in any site or splenomegaly and/or hepatomegaly (50% of presentations)
-
High risk (formerly stages III and IV) – Lymphocytosis with disease-related anemia (hemoglobin < 11 g/dL) or thrombocytopenia (platelets < 100 × 109/L) (25% of all patients)
The Binet staging system categorizes patients according to the number of lymph node groups involved. The areas of lymph node involvement considered are the head and neck, including the Waldeyer ring; axillae; groins; a palpable spleen; and a palpable liver. [2]
-
Stage A – Hemoglobin 10 g/dL or higher, platelets 100 × 10 9/L or higher, and up to two of the above areas involved.
-
Stage B – Hemoglobin and platelet levels as in stage A and three or more of the above lymph node areas involved
-
Stage C – Hemoglobin less than 10 g/dL and/or platelets less than 100 × 10 9/L
See Workup for more detail.
Management
Patients with early-stage CLL, including low Binet or Rai stages, are not treated with chemotherapy until they become symptomatic or display evidence of rapid progression of disease. Early initiation of chemotherapy has failed to show survival benefit in CLL. [2]
Although combination chemotherapy regimens, including the nucleoside analogue fludarabine, were once the most commonly used first-line therapy in CLL, non-chemotherapy regimens (eg, with Bruton tyrosine kinase [BTK] inhibitors) are currently preferred in most cases. Treatment selection takes into account the molecular and genetic characteristics of the disease and may include the following agents, as monotherapy or in combination [5] :
-
Acalabrutinib
-
Obinutuzumab
-
Venetoclax
-
Ibrutinib
-
Zanubrutinib
-
Rituximab (in combination therapies)
Fludarabine, cyclophosphamide, and rituximab (FCR) was previously a recommended first-line treatment for fit, young patients (< 60 years). [6, 7, 4] However, a 2019 clinical trial called this recommendation into question; it showed superior progression-free and overall survival with ibrutinib-rituximab compared with FCR, in particular in patients without the IGHV mutation, and lower rates of serious infectious complications with ibrutinib-rituximab. [8]
Allogeneic stem cell transplantation is the only known curative therapy for CLL. It should be discussed with patients as a treatment option at the point of first or second relapse. [2]
See Treatment and Medication for more detail.
For patient education information, see Leukemia and Living With Chronic Lymphocytic Leukemia.
Pathophysiology
The cells of origin in most cases of CLL are clonal B cells arrested in the B-cell differentiation pathway, intermediate between pre-B cells and mature B cells. Morphologically, in the peripheral blood, these cells resemble mature lymphocytes.
CLL B-lymphocytes typically show B-cell surface antigens, as demonstrated by CD19, CD20dim, CD21, and CD23 monoclonal antibodies. In addition, they express CD5, which is more typically found on T cells. Because normal CD5+ B cells are present in the mantle zone of lymphoid follicles, B-cell CLL is most likely a malignancy of a mantle zone–based subpopulation of anergic self-reactive cells devoted to the production of polyreactive natural autoantibodies.
CLL B-lymphocytes express extremely low levels of surface membrane immunoglobulin, most often immunoglobulin M (IgM) or IgM/IgD and IgD. Additionally, they also express extremely low levels of a single immunoglobulin light chain (kappa or lambda).
An abnormal karyotype is observed in the majority of patients with CLL. The most common abnormality is deletion of 13q, which occurs in over 50% of patients. Individuals showing 13q14 abnormalities have a relatively benign disease that usually manifests as stable or slowly progressive isolated lymphocytosis.
Deletion of 11q and 17p as well as trisomy 12 have also been reported, albeit less commonly. The presence of trisomy 12, which is observed in 15% of CLL patients, is associated with atypical morphology and progressive disease. Deletion in the short arm of chromosome 17 has been associated with rapid progression, short remission, and decreased overall survival. 17p13 deletions are associated with loss of function of the tumor suppressor gene p53. Deletions of bands 11q22-q23, observed in 19% of patients, are associated with extensive lymph node involvement, aggressive disease, and shorter survival [2] .
More sensitive techniques have demonstrated abnormalities of chromosome 12. About 40-50% of patients demonstrate no chromosomal abnormalities on conventional cytogenetic studies. However, 80% of patients will have abnormalities detectable by fluorescence in situ hybridization (FISH). Approximately 2-5% of patients with CLL exhibit a T-cell phenotype.
The proto-oncogene Bcl2 is overexpressed in B-cell CLL. [9] The proto-oncogene Bcl2 is a known suppressor of apoptosis (programmed cell death), resulting in a long life for the involved cells. Despite the frequent overexpression of Bcl-2 protein, genetic translocations that are known to result in the overexpression of Bcl2, such as t(14;18), are not found in patients with CLL. Analysis of the molecular pathophysiology of CLL allows the development of therapies that target leukemic cells with these genetic changes. Venetoclax, which is one of the first-line treatment options for CLL, is designed to block the Bcl2 protein.
Studies have shown that this upregulation in Bcl2 is related to deletions of band 13q14. Two genes, named miRNA15a and miRNA16-1, are located at 13q14 and have been shown to encode not for proteins, but rather for a regulatory RNA called microRNA (miRNA). [10, 11] These miRNA genes belong to a family of highly conserved noncoding genes throughout the genome whose transcripts inhibit gene expression by causing degradation of mRNA or by blocking transcription of mRNA.
Deletions of miRNA15a and miRNA16-1 lead to overexpression of Bcl2 through loss of downregulating miRNAs. Genetic analyses have demonstrated deletion or downregulation of these miRNA genes in 70% of CLL cases. [12]
A study of the CLL genome by Wang et al discovered splicing factor 3b (SF3B1) mutations affecting pre-mRNA in 15% of sampled cells. [13] SF3B1 mutations are also found in 20% of myelodysplastic syndrome cases. [14] In future this may provide a therapeutic target.
Investigations have also identified a number of high-risk genetic features and markers, including the following:
-
Germline immunoglobulin variable heavy chain (IgV H)
-
IgV H V3-21 gene usage
-
Increased CD38 expression
-
Increased Zap70 expression
-
Elevated serum beta-2-microglobulin levels
-
Increased serum thymidine kinase activity
-
Short lymphocyte doubling time (< 6 months)
-
Increased serum levels of soluble CD23
These features have been associated with rapid progression, short remission, resistance to treatment, and shortened overall survival in patients with CLL.
Germline IgVH has been shown to indicate a poor prognosis. Studies have shown that these patients also have earlier progression of CLL after treatment with chemotherapy. Zeta-associated peptide of 70 kilodaltons (Zap70) is a cytoplasmic tyrosine kinase whose expression has also been associated with a poor prognosis. Cells with germline IgVH often have an increased expression of Zap70; however, studies have shown discordance rates of 10-20% between IgVH mutational status and Zap70 expression levels. Elevated levels of Zap70 are believed to decrease the threshold for signaling through Bcl2, thereby facilitating the antiapoptotic effects of Bcl2.
Etiology
As is the case with most malignancies, the exact cause of CLL is uncertain. CLL is an acquired disorder, and reports of truly familial cases are exceedingly rare. [15] A meta-analysis of four genome-wide association studies that included 3100 cases of CLL found multiple risk loci. Several of those loci are in close proximity to genes involved in apoptosis, suggesting a plausible underlying biological mechanism. [16]
Epidemiology
United States Statistics
The American Cancer Society estimates that 18,740 new cases of CLL will be diagnosed in the United States in 2023. [17] The true incidence in the US is unknown and is likely higher, as estimates of CLL incidence come from tumor registries, and many cases are not reported. Although the incidence of CLL has been stable over the last two decades, mortality has been steadily declining. [2]
International Statistics
Although the incidence of CLL in Western countries is similar to that of the United States, CLL is extremely rare in Asian countries (eg, China, Japan), where it is estimated to comprise only 10% of all leukemias. However, underreporting and incomplete registries may significantly underestimate the true incidence of CLL in those countries.
Race-, sex-, and age-related demographics
The incidence of CLL is higher in Whites than in Blacks. The incidence of CLL is higher in males than in females, with a male-to-female ratio of 1.9:1. [2] CLL is a disease that primarily affects the elderly, with the median age at diagnosis being 70 years. [18] In familial CLL (ie, disease in patients with at least one first-degree relative with CLL), median age at diagnosis is 57 years. [19]
Prognosis
The prognosis of patients with CLL varies widely at diagnosis. Some patients die rapidly, within 2-3 years of diagnosis, because of complications from CLL. In most patients CLL initially has a relatively benign course, but eventually enters a progressive, treatment-resistant phase. During this later phase, morbidity is considerable, both from the disease and from complications of therapy. [20, 21]
With the advances in treatment of CLL in recent decades, prolonged survival is possible. However, treatment rarely cures CLL, and mortality rates in patients with CLL remain significantly higher than in the general population. [22]
Prognosis depends on the disease stage at diagnosis as well as the presence or absence of high-risk markers (see Pathophysiology). Given the recent advancements in CLL treatment, the Rai and Binet staging systems do not provide sufficient utility to estimate prognosis. The most accurate prognostic score currently utilized by oncologists is the CLL International Prognostic Index (CLL-IPI), which relies on five independent prognostic factors [2] :
- Patient age
- Clinical stage (Rai or Binet)
- Serum B2 microglobulin level
- Mutational status of immunoglobulin heavy chain variable (IGVH)
- 17p deletion and/or TP53 mutation
Patients are deemed low-, intermediate-, high-, or very high risk depending on the presence or absence of the prognostic factors (see the CLL-IPI calculator). Low-risk patients have over a 90% chance of 5-year overall survival and, therefore, treatment is not recommended. Intermediate-risk patients have a nearly 90% chance of 5-year overall survival and are treated only if they are symptomatic. High-risk patients are managed similarly to intermediate-risk patients, but they have a slightly lower chance of 5-year overall survival at 63%. Very high-risk patients are treated with targeted agents (see Treatment and Medication.
-
Peripheral smear from a patient with chronic lymphocytic leukemia, small lymphocytic variety.
-
Peripheral smear from a patient with chronic lymphocytic leukemia, large lymphocytic variety. Smudge cells are also observed; smudge cells are the artifacts produced by the lymphocytes damaged during the slide preparation.
Tables
| CLL-IPI category | Overall Survival at 5 years | Treatment Recommendation |
|---|---|---|
| Low risk | 93.2% | Do not treat |
| Intermediate risk | 79.3% | Treat only if symptomatic |
| High risk | 63.3% | Treatment indicated; however, can consider no treatment if asymptomatic |
| Very high risk | 23.3% | If treatment needed, do not use chemotherapy; instead, use targeted agent or clinical trial |







