Practice Essentials
Idiopathic pulmonary arterial hypertension (IPAH) is a rare disease characterized by elevated pulmonary artery pressure with no apparent cause. IPAH is also termed WHO Group I pulmonary hypertension (PH), precapillary pulmonary hypertension, and, previously, primary pulmonary hypertension. Untreated, IPAH leads to right-sided heart failure and death.
Often, the first sign that PH exists is an abnormal echocardiogram. Typically, the echocardiogram demonstrates evidence of increased pulmonary arterial (PA) pressure, accompanied by an enlarged right ventricle (see the image below).
The echocardiogram may also demonstrate the presence of a pericardial effusion. In approximately a third of patients with pulmonary arterial hypertension (PAH), Doppler echocardiography demonstrates right-to-left shunting across a patent foramen ovale.
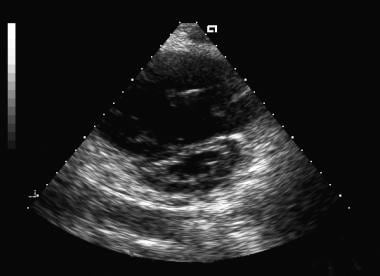 Two-dimensional short-axis echocardiogram image. Note the flattened interventricular septum due to right ventricular overload.
Two-dimensional short-axis echocardiogram image. Note the flattened interventricular septum due to right ventricular overload.
Signs and symptoms
Symptoms of IPAH are nonspecific and commonly include the following:
-
Dyspnea
-
Fatigue
-
Weakness
-
Syncope or presyncope
Cardiovascular examination in patients with PAH often reveals the following findings:
-
The pulmonic component of the second heart sound is usually increased, which may demonstrate fixed or paradoxic splitting in the presence of severe right ventricular dysfunction; occasionally, the second heart sound may be palpable
-
A pulmonic regurgitation (Graham Steell murmur) may be apparent
-
A murmur of tricuspid regurgitation can be present, and a right ventricular lift (heave) may be noted
-
Jugular venous pulsations may be elevated in the presence of volume overload, right ventricular failure, or both; large V waves are often present because of the commonly present severe tricuspid regurgitation, and large a waves thought to be secondary to poor right ventricular compliance may also be present
-
Right-sided S3 gallop
Other findings may include the following:
-
Hepatomegaly with palpable pulsations of the liver
-
Abnormal abdominal-jugular reflex
-
Ascites - Not uncommon in untreated patients and in patients with worsening decompensated right heart failure
-
Pitting edema - In the extremities
-
Presacral edema - In patients who are bedridden
See Clinical Presentation for more detail.
Diagnosis
Cardiac catheterization
Cardiac catheterization is the criterion standard test to definitively confirm any form of PAH. It is essential in the workup of all patients suspected of IPAH. Excluding left-sided heart disease, including diastolic dysfunction, is especially important in these patients because of major treatment implications. Catheterization is also performed to determine pulmonary vasoreactivity, which can be prognostic and figures in the initiation and titration of high-dose calcium channel blocker (CCB) therapy.
Laboratory studies
-
Antinuclear antibody
-
Thyroid function
-
B-type natriuretic peptide, or N-terminal B-type natriuretic peptide
-
Pulmonary function testing (PFT)
Imaging studies - Essential
-
Chest radiography: A chest radiograph may help identify secondary causes of, or contributors to, pulmonary hypertension.
-
Echocardiography: The echocardiogram can estimate right ventricular (RV) systolic pressure and, more importantly, the degree of right ventricular enlargement, as well as the presence of a pericardial effusion. Equally important, the echocardiogram helps to exclude secondary causes of, or contributors to, pulmonary hypertension, such as left-sided heart disease (eg, left ventricular dysfunction, valvular heart disease).
-
Nuclear lung ventilation/perfusion scanning: This is performed to exclude chronic thromboembolic pulmonary hypertension (Group IV PH).
Imaging studies - Conditional
-
Chest computed tomography (CT)
-
Magnetic resonance imaging (MRI)
-
Pulmonary angiography
Electrocardiography
Electrocardiographic results are often abnormal in patients with PAH, revealing right atrial enlargement, right axis deviation, right ventricular hypertrophy, and characteristic ST depression and T-wave inversions in the anterior leads. Sometimes, an incomplete RBBB may be seen (usually in patients with atrial septal defects). However, some patients with IPAH have few or no abnormal electrocardiographic findings.
Histology
Several histologic subtypes are associated with pulmonary arteriopathy in IPAH, including plexogenic arteriopathy, in situ thrombosis, and intimal and adventitial proliferation.
Exercise testing
In patients with IPAH, values for peak exercise oxygen consumption, oxygen pulse, and ventilator equivalents (ratio of expired volume to carbon dioxide output [ie, wasted ventilation fraction] at the anaerobic threshold) during exercise are abnormal to varying degrees.
Commonly, a 6-minute walk test is performed in the office as a crude measurement of exercise capacity.
See Workup for more detail.
Management
Calcium channel blocker therapy
Only in a small subset of IPAH patients, long-term treatment results in clinical improvement; this is in patients who are proven responders to calcium channel blockers (CCBs) and do not have contraindications to CCBs (depressed cardiac output and/or systemic hypotension). In general, CCBs are used at high doses in patients with IPAH.
PAH-specific therapy
For patients with IPAH in whom CCBs are contraindicated (most IPAH patients), or in whom CCBs are ineffective or poorly tolerated, guidelines from the joint European Respiratory Society (ERS)/European Society of Cardiology (ESC) recommend using the patient’s World Health Organization (WHO) functional class to guide the choice of PAH-specific therapy. [1]
More comprehensive guidelines encompass several clinical parameters that are used to determine risk of adverse outcomes. These include functional class, exercise capacity, symptom progression rate, the presence or absence of heart failure on examination, certain biomarkers, and findings on echocardiography. [1]
Transplantation and septostomy
-
Lung transplantation - A single- or double-lung transplant is indicated for patients who do not respond to medical therapy
-
Septostomy - Atrial septostomy is a palliative procedure that may afford some benefit to patients whose condition is deteriorating
See Treatment and Medication for more detail.
Background
Idiopathic pulmonary arterial hypertension (IPAH) is a rare disease characterized by elevated pulmonary artery pressure with no apparent cause. IPAH is also termed precapillary pulmonary hypertension and was previously termed primary pulmonary hypertension. The term IPAH is now the preferred term for pulmonary arterial hypertension of unknown etiology; thus, IPAH represents pulmonary vascular disease with a spectrum of clinical presentations.
A complete classification of all pulmonary hypertension (PH) types has been updated [2] :
1. Pulmonary arterial hypertension (PAH)
1.1 Idiopathic PAH
1.2 Heritable PAH
1.2.1 BMPR2
1.2.2 ALK-1, ENG, SMAD9, CAV1, KCNK3
1.2.3 Unknown
1.3 Drug- and toxin-induced
1.4 Associated with:
1.4.1 Connective tissue disease
1.4.2 HIV infection
1.4.3 Portal hypertension
1.4.4 Congenital heart disease
1.4.5 Schistosomiasis
1'. Pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis
1". Persistent pulmonary hypertension of the newborn (PPHN)
2. Pulmonary hypertension due to left heart disease
2.1 Left ventricular systolic dysfunction
2.2 Left ventricular diastolic dysfunction
2.3 Valvular disease
2.4 Congenital/acquired left heart inflow/outflow tract obstruction and congenital cardiomyopathies
3. Pulmonary hypertension due to lung diseases and/or hypoxia
3.1 Chronic obstructive pulmonary disease
3.2 Interstitial lung disease
3.3 Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4 Sleep-disordered breathing
3.5 Alveolar hypoventilation disorders
3.6 Chronic exposure to high altitude
3.7 Developmental lung diseases
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. Pulmonary hypertension with unclear multifactorial mechanisms
5.1 Hematologic disorders: chronic hemolytic anemia, myeloproliferative disorders, splenectomy
5.2 Systemic disorders: sarcoidosis, pulmonary histiocytosis, lymphangioleiomyomatosis
5.3 Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4 Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure, segmental PH
Within this classification, IPAH represents a subset of pulmonary vascular disease called pulmonary arterial hypertension (Group I PH, or PAH), which includes conditions known to be associated with pulmonary hypertension that share similar pathophysiology to IPAH. Conditions in which PAH and these associated conditions co-exist are called associated PAH (APAH).
Dresdale and colleagues first reported a hemodynamic account of IPAH in 1951. [3] However, the pathophysiology of IPAH remains poorly understood. At least 15-20% of patients previously thought to have IPAH actually have a familial (heritable) form of PAH involving at least one genetic defect, which has only recently been characterized (see Pathophysiology).
By definition, pulmonary hypertension refers to the condition in which resting mean pulmonary arterial pressure (mPAP) is greater than 25 mmHg. Further, in order to hemodynamically distinguish PAH (IPAH and APAH) from other forms of PH, the pulmonary capillary wedge (PCW) pressure must be less than 15 mmHg, and the pulmonary vascular resistance (PVR) must be more than 3 Wood Units. Thus, cardiac catheterization is the criterion standard test to definitively confirm any form of PAH, including IPAH. However, a thorough workup includes a range of additional testing to exclude all reasonable causes of secondary pulmonary hypertension (see Workup).
Until recently, calcium channel blockers (CCBs) had been the most widely used class of drugs for IPAH. Patients with IPAH in whom CCBs are contraindicated, ineffective, or poorly tolerated may respond to long-term PAH-specific therapy (see Treatment and Management).
Treating IPAH requires significant knowledge of and exposure to the available therapies for IPAH and their potential complications. Because IPAH is relatively rare, management is best left to expert personnel at centers with regular exposure to these patients (see Treatment and Management).
Also see Pediatric Idiopathic Pulmonary Artery Hypertension.
Pathophysiology
The pathophysiology of IPAH is poorly understood. An insult (eg, hormonal, mechanical, other) to the endothelium may occur, possibly in the setting of increased susceptibility to pulmonary vascular injury (ie, multiple hit theory), resulting in a cascade of events characterized by vascular scarring, endothelial dysfunction, and intimal and medial (smooth muscle) proliferation.
At least 15-20% of patients previously thought to have IPAH actually have a familial form of PAH involving at least one genetic defect. The most common genetic defect in these cases involves the BMPR-II gene. However, only about a third of affected patients with a family history of PAH have an identifiable BMPR-II mutation. This suggests that additional genetic abnormalities and/or additional external factors may exist that predispose individuals to developing PAH.
In 2013, 6 mutations that appear to be associated with PAH and that may be treatable with PAH drugs were discovered in a gene, KCNK3, that had not previously been linked to the disease. Each of the 6 mutations was linked to a loss of function of potassium ion channels. [4, 5] In vitro examination of the investigational agent ONO-RS-082 (2-[p-amylcinnamoyl]amino-4-chlorobenzoic acid), a phospholipase A2 inhibitor, found that for 2 of the 3 mutations tested, the drug restored function to nonworking potassium ion channels.
The current Nice Classification system of PH now lists the following genetic defects that are known to be associated with PAH [6] :
-
BMPR2
-
ALK1
-
ENG
-
SMAD9
-
CAV1
-
KCNK3
Early in IPAH (and probably in APAH), as the pulmonary artery pressure increases because of increasing right ventricle work, thrombotic pulmonary arteriopathy occurs. Thrombotic pulmonary arteriopathy is characterized by in situ thrombosis of small muscular arteries. In later stages, as the pulmonary pressure continues to rise, plexogenic pulmonary arteriopathy develops. This is characterized by a remodeling of the pulmonary vasculature with intimal fibrosis and replacement of normal endothelial structure.
For more information, see the Medscape Reference article Persistent Newborn Pulmonary Hypertension.
Associated conditions
Pulmonary vascular disease can be associated with portal hypertension (sometimes called portopulmonary hypertension), suggesting that patients with shunting of splanchnic blood, with or without liver disease, have a higher risk of developing PAH.
Additionally, exposure of the pulmonary circulation to substances from the splanchnic circulation that are normally detoxified via the liver may contribute to the development of pulmonary hypertension. More research is necessary to better understand this relationship.
Patients with connective-tissue diseases, namely the CREST (calcinosis cutis, Raynaud phenomenon, esophageal motility disorder, sclerodactyly, and telangiectasia) variant of scleroderma, systemic lupus erythematosus, and mixed connective-tissue disease, are also predisposed to developing IPAH-like disease. This is now termed associated PAH, or APAH.
The pathophysiologic nature of this predisposition is unclear. In the past, most experts used the term "secondary" pulmonary arterial hypertension for these diseases, indicating that, similar to IPAH, the process involves the precapillary circulation but is somehow caused by or at least associated with the underlying (predisposing) disease.
A study by Soon et al determined that unexplained iron deficiency is more prevalent in patients with idiopathic pulmonary artery hypertension than in those with chronic thromboembolic pulmonary hypertension (CTEPH). [7] Interleukin-6 (IL-6) may play a role in this difference in prevalence.
Etiology
The strict definition of IPAH is pulmonary hypertension with no known cause. However, associations have been recognized (eg, connective-tissue diseases, liver cirrhosis, exposure to anorexigens and likely other alpha-adrenergic stimulants [eg, cocaine, amphetamines], [8] HIV infection). How these associated conditions predispose to or cause PAH remains unknown.
Epidemiology
IPAH is responsible for approximately 125-150 deaths per year in the United States and has an incidence rate of approximately 2-6 cases per million population per year. The incidence and prevalence of APAH are considerably higher than those of IPAH. The worldwide incidence of IPAH approximates that observed in the United States, but variations in prevalence exist worldwide. A registry of patients with IPAH in France found a prevalence of IPAH of about 6 cases per million population. [9] IPAH occurs at a female-to-male ratio ranging from 2-9:1, depending on the treatment center sampled. In the United States, the average female-to-male ratio reported in clinical trials and registries is close to 4:1. The reasons for this female predilection remain unknown.Typically, younger women of childbearing age develop IPAH. However, IPAH can also affect individuals in their fifth and sixth decades of life or older. [10]
Prognosis
IPAH has no cure. Untreated IPAH leads to right-sided heart failure and death. Prior to the 1990s, therapeutic options were limited. The emergence of prostacyclin analogues, endothelin receptor antagonists, phosphodiesterase-5 inhibitors, and other novel drug therapies has greatly improved the outlook for patients with IPAH and IPAH-like diseases.
For untreated IPAH, the estimated 3-year survival rate is approximately 41%. In one study of long-term continuous intravenous prostacyclin therapy, 3-year survival increased to approximately 63%. [11] With newer therapies, perhaps in combination, these figures are expected to improve further.
Data on long-term survival in patients treated with other pulmonary vascular therapies are emerging. Patients whose disease progresses and is unresponsive to medical treatments either undergo transplantation or die of progressive right-sided heart failure.
Using the largest registry of patients with PAH to date, the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL Registry), Benza et al analyzed factors determining survival in 2716 patients. [12] Using this data, they derived a multivariable, weighted risk formula incorporating 19 independent factors identified as having an impact on PAH patient survival, thus allowing clinicians to incorporate factors encountered in real-world management of PAH in their overall risk/severity assessment.
In another analysis of data from the REVEAL Registry, Frost et al found that PAH patients with mean pulmonary capillary wedge pressure (PCWP) of 16-18 mmHg at diagnostic right heart catheterization were heavier, older, and were more likely to have comorbidities associated with left ventricular diastolic dysfunction at diagnosis than patients with PCWP ≤15 mmHg. Five-year survival was poor in both PCWP subgroups. [13]
Patient Education
Patient education about this rare fatal disease is paramount. If applicable, instruct patients on how to administer their daily parenteral medication. For patient education information, see the Lung Disease & Respiratory Health Center and Heart Health Center.
-
Two-dimensional short-axis echocardiogram image. Note the flattened interventricular septum due to right ventricular overload.
Tables

What would you like to print?
- Overview
- Presentation
- DDx
- Workup
- Treatment
- Guidelines
- Medication
- Medication Summary
- Calcium Channel Blockers
- Parenteral Prostanoids
- PAH, PDE-5 Inhibitors
- Inhaled Prostanoids
- Oral Prostacyclin Analogues
- Prostacyclin Receptor Agonists
- Endothelin Receptor Antagonists
- Soluble Guanylate Cyclase (sGC) Stimulators
- Activin Signaling Inhibitors
- Diuretics
- Anticoagulants
- Cardiac Glycosides
- Show All
- Questions & Answers
- References







