Practice Essentials
Gonadotropin-releasing hormone (GnRH) is a neurohormone central to initiation of the reproductive hormone cascade. Pulsatile secretion of GnRH from the hypothalamus is key in establishing and maintaining normal gonadal function. Failure of this release results in isolated GnRH deficiency that can be distinguished by partial or complete lack of GnRH–induced luteinizing hormone (LH) pulses, normalization with pulsatile GnRH replacement therapy, and otherwise normal hypothalamic-pituitary neuroanatomy and neurophysiology.
Signs and symptoms
Most physical findings are related to failure of sexual maturation. Affected patients have eunuchoidal body habitus, with arm-span greater than height by 5 cm or more. Secondary sexual characteristics are often absent. Women have little or no breast development, and men have little or no facial hair.
See Presentation for more detail.
Diagnosis
Laboratory studies
Most patients have low serum levels of basal gonadotropins, estrogen/testosterone, and poor response to GnRH stimulation. The GnRH stimulation test using a synthetic GnRH analog, such as buserelin, has been used to differentiate males with gonadotropin deficiency from those with delayed puberty.
Imaging studies
Magnetic resonance imaging appears to be the single best study for the diagnosis of Kallmann syndrome and exclusion of other central nervous system disorders associated with hypogonadotropic hypogonadism.
See Workup for more detail.
Management
The choice of therapy depends on the patient's desire to achieve one or more of the following:
-
Secondary sex characteristics
-
Fertility
-
Bone and muscle mass
See Treatment and Medication for more detail.
Background
Clinicians and scientists have long been intrigued by the findings of olfactory disturbances and concomitant reproductive dysfunction. In 1856, Spanish pathologist Maestre de San Juan noted the association between the failure of testicular development and the absence of the olfactory bulbs. However, the syndrome comprising complete GnRH deficiency and lack of olfactory senses is named Kallmann syndrome (KS) after the American geneticist Kallmann.
In 1944, Kallmann, Schoenfeld, and Barrera were the first to identify a genetic basis to this disorder. [1, 2] In 1954, de Morsier connected the syndrome of hypogonadism and anosmia with the abnormal development of the anterior portion of the brain. [3] KS is a rare disorder that occurs in both sexes. In contrast to KS, GnRH deficiency leading to hypogonadotropic hypogonadism with an intact sense of smell is termed idiopathic hypogonadotropic hypogonadism (IHH). IHH results from dysfunction of GnRH neurons that have developed and migrated properly, whereas KS is caused by defective migration of GnRH neurons to their proper position in the hypothalamus during fetal development. [4]
Pathophysiology
Gonadotropin-releasing hormone neurons
A fundamental understanding of the anatomy, biochemistry, ontogeny, and physiology of GnRH neurons aids in understanding the pathophysiology, diagnosis, and treatment of KS and idiopathic hypogonadotropic hypogonadism (IHH).
Gonadotropin-releasing hormone and gonadotropin-releasing hormone receptors
The decapeptide GnRH is derived from posttranslation processing of a tripartite 92–amino acid (AA) pre-pro-GnRH. The first 23 AA is a signal peptide and the last 56 AA is known as GnRH–associated protein (GAP). GnRH is encoded from a single gene located on the short arm of chromosome 8. Serum levels of GnRH are difficult to obtain due to its short half-life (2-4 min) and complete confinement to the hypophyseal-portal blood supply. Due to the small structure and ease of mutation of GnRH, chemists have created several clinically useful GnRH analogs. GnRH binds with high affinity to cell surface LH and follicle stimulating hormone (FSH) receptors located on the pituitary gonadotrophs. These 7-transmembrane, cell surface G protein-coupled receptors activate phospholipase C (PLC).
PLC leads to the activation of several second messenger molecules, the most important being diacylglycerol (DG) and inositol 1,4,5-trisphosphate (IP3). In turn, DG activates protein kinase C and causes IP3 -stimulated release of calcium ions from intracellular pools. The result is the synthesis and release of FSH and LH from the pituitary gonadotrophs. The released gonadotropins stimulate the gonads to produce steroid hormones, and in the testes, to produce sperm or in the ovaries, to release oocytes. Mutated GnRH receptors (GnRH-R), as predicted by the biochemistry, could result in the clinical manifestations of isolated gonadotropin deficiency. Many factors interact to regulate the synthesis and secretion of GnRH, and to regulate the translation of GnRH receptors; the review of this regulation is beyond the scope of this article.
Ontogeny and function
During fetal development, the migration of GnRH neurons follows a precise path from the olfactory placode to the preoptic area of the hypothalamus in mammals. The olfactory placode is composed of thickened ectoderm that is lateral to the head of the developing embryo and invaginates to form simple olfactory pits on either side of the nasal septum. The lateral epithelium of the olfactory pits gives rise to the olfactory nerves. The medial portion develops into the site of initial GnRH appearance and the terminal nerves. The terminal nerves, ganglionated cranial nerves for which the exact function is unknown, enter the forebrain and serve as a highway for the GnRH neuronal migration. In humans, GnRH neuron migration begins in the 6th week of embryonic development.
Migrating GnRH neurons do not contain neurosecretory vesicles until they reach the area of the arcuate nucleus in the hypothalamus. For this reason, neurons that do not reach the forebrain are unable to secrete GnRH. GnRH neurons have been identified in the fetal hypothalamus at 9 weeks' gestation and are connected to the pituitary portal system by 16 weeks' gestation. At 10 weeks' gestation, gonadotropes are detectable in the pituitary, and by the 12th week, FSH and LH are measurable in the bloodstream. Fetal peripheral blood levels of gonadotropins peak during the second trimester of pregnancy and decrease by term as the negative feedback mechanism develops.
LH pulsatility, which can be measured in the bloodstream, is determined by the precise frequency and amplitude of pulsatile GnRH release; thus, serum LH is used as a marker of GnRH pulsatility. [5]
GnRH is secreted during the neonatal period, resulting in pulsatile LH and FSH secretion, which decreases by age 6 months in boys and by age 1-2 years in girls until puberty. Before the initiation of puberty, GnRH is still secreted in a pulsatile fashion but at reduced amplitude and frequency. The hypothalamic pulse generator, the master regulator of GnRH secretion, is likely suppressed by a mechanism that inhibits GnRH release but not its synthesis.
This theory has been demonstrated in primates, in which GnRH messenger RNA (mRNA) and proteins are abundant in the hypothalamus during an equivalent developmental stage.
The pubertal period is characterized by a predominantly nocturnal increase in both the amplitude and frequency of GnRH–induced LH pulses. Sex steroids are secreted from the gonads in response to this nocturnal increase in gonadotropins. Gonadotropins continue to be secreted in a pulsatile fashion, under the control of pulsatile GnRH release, during adulthood. The mechanism that awakens the pubertal surge of more robust GnRH secretion is not completely understood. Metabolic cues, steroid hormones, neurosteroids, growth factors, and neurotransmitter systems have been implicated, including glutamate, gamma-aminobutyric acid, neuropeptide Y (NPY), opioids, leptin, kisspeptin, and estradiol. [6]
Most studies in males have shown LH pulses to occur every 2 hours; in females, LH (and thus GnRH) pulse frequency varies throughout the menstrual cycle. In the early follicular phase, LH pulse frequency is every 90 minutes and increases to every 60 minutes by the late follicular phase. The LH "surge" that triggers ovulation occurs due to a "switch" from negative to positive feedback of estrogen at the pituitary, leading to a brief burst of pulsatile LH release, which stimulates ovulation. [7] Following ovulation, LH pulse frequency decreases, with frequency ranging from every 4-8 hours during the luteal phase.
Studying gonadotropin-releasing hormone secretion
Studying GnRH physiology in humans and animal models has been challenging. GnRH itself is almost entirely confined to the portal blood supply of the pituitary, thus direct sampling in humans is not feasible, and difficult if not impossible in animal models. Measurements of GnRH in the periphery are inaccurate because of its rapid 2-minute to 4-minute half-life. Much of the information known about GnRH has come from animal studies.
Belchetz and coworkers in the 1970s demonstrated in rhesus monkeys that pulsatile release of GnRH is required for maintaining gonadotrope function. [8] The researchers were able to differentiate between episodic and continuous stimulation by GnRH causing maintenance and desensitization, respectively, of the gonadotrope response.
Another model developed to study GnRH neuron function is immortalized GnRH cell lines. Interestingly, implantation of these cells into the hypothalami of female GnRH–deficient mice restores normal estrus (equivalent of menstrual) cycles. Immortalized GnRH cell lines in culture have provided an important in vitro tool for studying reproductive neuroendocrine function. In vivo studies of GnRH neuron function have also been possible since development of transgenic mouse models in which GnRH neurons are labeled with green fluorescent protein (GnRH-GFP mouse). [9] This model allows GnRH neurons to be visualized in vivo in hypothalamic sections. Studies from this model are elucidating the complex physiology of GnRH neurons, including neuronal firing patterns, neuronal inputs, migratory patterns, and intracellular signaling systems.
Human studies have been limited to frequent sampling studies in healthy and diseased models, the use of pharmacological probes, and genetic studies. As in animals, LH has long been used as a marker of GnRH pulse activity in humans. Most recently, the glycoprotein free alpha subunit (FAS) has been used as a marker due to its correlation with LH. FAS is useful in tracking GnRH because of its 12-minute to 15-minute half-life. In addition to LH and FAS, an estimate of endogenous GnRH can be obtained using GnRH antagonists as probes. Administering a GnRH antagonist induces a GnRH receptor blockade so that the amount of GnRH present is inversely proportional to the amount of LH inhibitor.
Etiology
GnRH deficiency is inherited through autosomal dominant, autosomal recessive, and X-linked transmissions. However, more than two thirds of cases are sporadic. In fact, only 30% of cases of GnRH deficiency are due to mutations in known genes.
Evidence suggests that most familial cases of GnRH deficiency are controlled by autosomal inheritance. In a study of 106 patients with GnRH deficiency at Massachusetts General Hospital, only 21% of familial cases were X-linked. [10] Using isolated congenital anosmia as a marker for KS, X-linked and autosomal recessive transmission was 18% and 32%, respectively. Autosomal dominance accounted for 50% of cases. When delayed puberty was included in the phenotypic analysis, X-linked cases accounted for 11% of cases, whereas autosomal recessive and autosomal dominant cases were 25% and 64%, respectively.
KAL1 gene
The KAL1 gene, described in 1991, is an example of an X-linked gene that encodes anosmin 1, an extracellular glycoprotein that is similar in amino acid structure to molecules involved in neural development, such as protease inhibitors, neurophysins, and neural cell adhesion molecules. [11] Anosmin 1 appears to be important to the migration of the GnRH neurons to their resting place in the hypothalamus. The KAL1 gene is located on the short arm of the X chromosome at Xp22.3. Approximately 10-20% of males with KS have KAL1 gene mutations, and the phenotypes associated with this mutation tends to be more severe and less variable compared to other KS mutations. KAL1 mutations are inherited in an X-linked recessive pattern and produce a syndrome of short stature, intellectual disability, ichthyosis, chondroplasia punctata, and KS.
Most of the data on the KAL1 gene come from studies in chickens. The timing of KAL1 expression in the chicken has aided in understanding the migration defects of GnRH neurons in human KS. KAL1 is expressed in 2 distinctly different periods of embryonic development. KAL1 expression is found in limb buds, facial mesenchyme, and the neurons innervating the extrinsic eye muscles during embryonic development. By embryonic day 5 (of a 21 day incubation period of a chicken), GnRH neurons migrate along the olfactory nerve and penetrate the olfactory bulb by embryonic day 7-8. KAL1 expression is increased in the olfactory bulb by embryonic day 7-8. At embryonic day 9-10, KAL1 expression is up-regulated as synapses are formed between the olfactory nerve and the mitral cell layer.
Studies have demonstrated that neural migration is controlled by factors intrinsic to the olfactory epithelium. When the olfactory placode is destroyed in the chick, KAL1 expression continues in the olfactory bulb, suggesting that KAL1 expression and olfactory nerve innervation are independent of one another. In humans, KAL1 transcripts are not identified at the time of olfactory nerve migration, again suggesting independence between KAL1 expression and olfactory nerve migration. In KS, a defect in neuronal interaction, rather than neural migration, has been suggested. In a study of a 19-week fetus with X-linked KS, the olfactory nerves were shown to have arrested within the meninges, whereas the GnRH neurons were arrested in the forebrain, never reaching the hypothalamus. Both groups of neurons passed through the cribriform plate but arrested prematurely. The KAL1 gene may play a later role, such as controlling the penetration of GnRH neurons into the olfactory bulb.
Without KAL1 and without functioning synaptic connections, the olfactory nerve might atrophy and degenerate, causing the defective GnRH migration.
The KAL1 gene may also play a role in the development of other tissues, such as facial mesenchyme, fibrous and perichondral cells, blood vessels, renal glomeruli, and developing limb buds. In humans, defective KAL1 expression in the cerebellum may be linked to nystagmus and ataxia observed in some patients with KS.
Fibroblast growth factor receptor 1 and fibroblast growth factor 8
There are 2 KS-related loci, KAL1 and KAL2. The former encodes anosmin and is described above. KAL–2 encodes the fibroblast growth factor receptor 1 (FGFR1). Approximately 10% of patients with KS have loss-of-function mutations in FGFR1. [12] The KAL2- associated disorder is inherited in an autosomal dominant manner. The clinical phenotype ranges from severe KS to delayed puberty. [13] Associated features include cleft palate, hearing loss, agenesis of the corpus callosum, and fusion of metacarpal bones. In affected individuals, the lack of smell has a variable penetrance. [14] Anosmin, a product of KAL1 gene, interacts and enhances the signaling of FGFR1. [15] Thus, in FGFR1 heterozygous affected women, the KAL gene, by escaping X-inactivation, may rescue FGFR1 signaling. [16] This effect of X-inactivation likely explains why this condition is more prevalent in males.
In addition to FGFR1, fibroblast growth factor 8 (FGF8) gene mutations have also been associated with KS and IHH, with varying degrees of olfactory and reproductive function. [17] Interestingly, a mouse model of FGF8 deficiency lacks both hypothalamic GnRH neurons and olfactory bulbs, suggesting a role for FGF8 in olfactory and GnRH neuron migration. [18]
Prokineticin 2 and prokineticin 2 receptor genes
Prokineticin 2 (PROK2) and its receptor (PROKR2) are a ligand-receptor pair involved in the development of the olfactory bulbs and GnRH neuron migration. Neurogenesis persists in the olfactory bulb of the adult mammalian brain due to the chemoattractant effect of prokineticin 2 (PROK2). In PROK2 -deficient and PROKR2 -deficient mice, there is a significant reduction in olfactory bulb size and impaired neuronal migration. [19, 20] Mutations in PROK2 and in the receptor (PROKR2) gene have been associated with the development of KS and normosmic IHH, with variable phenotypic severities. [21, 22] In one series, 9% of patients with KS had mutations in either PROK2 or PROKR2. [23] Accompanying phenotypic features include fibrous dysplasia, synkinesia, and epilepsy.
G protein-coupled receptor 54
G protein-coupled receptor 54 (GPR54) binds to kisspeptin and its derivatives. This receptor is widely expressed throughout the brain. It has been shown that in a large consanguineous Saudi family with 6 individuals with IHH, a homozygous single nucleotide change in exon 3 of GPR54 was found in all 6 affected individuals, resulting in substitution of a serine for the normal leucine in the second intracellular loop of the receptor (L148S). See the image below.
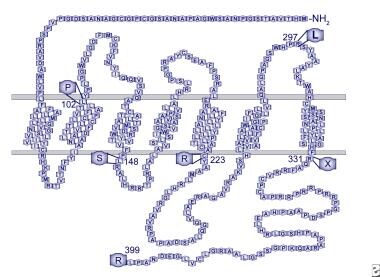 Human GPR54 receptor model. Mutations identified in patients with idiopathic hypogonadotropic hypogonadism are indicated.
Human GPR54 receptor model. Mutations identified in patients with idiopathic hypogonadotropic hypogonadism are indicated.
This change did not occur in the homozygous state in any unaffected family members and was not identified in any controls. This 7-transmembrane domain receptor shares highest homology, about 45%, with the galanin subfamily of receptors. The amino acid sequence is highly conserved across species, with 95% homology between the rat and mouse and 82% between mouse and human (98% in the transmembrane domains). [24]
A GPR54-deficient mouse model resulted in a phenotype similar to that seen in humans with KS. These mice have normal hypothalamic GnRH content, but develop IHH that is responsive to GnRH therapy, suggesting that. GnRH neurons continue to synthesize GnRH, but that GPR54 is necessary for GnRH processing and/or secretion. The ligand for GPR54 has been identified as the 54 amino acid protein metastatin. Kisspeptin, a 145-amino acid precursor, gives rise to metastin after cleavage. GPR54 activation advances puberty in rodents and overcomes amenorrhea that is due to starvation or leptin deficiency. Thus, the kisspeptin/metastin/GPR54 system is clearly a major gatekeeper of the pubertal process. [25] Furthermore, the kisspeptin/metastin/GPR54 system plays a major role in the sexual differentiation of the brain and sexual behavior. [26]
Of note, no mutations responsible for KS/IHH have been reported in the KISS1 gene, the gene encoding kisspeptin itself.
Gonadotropin-releasing hormone receptor and gonadotropin-releasing hormone 1
The GnRH receptor is a G protein–coupled receptor, which activates phospholipase C, ultimately mobilizing intracellular calcium. Mutations in the GnRH receptor (GnRHR) have been described in families with hypogonadotropic hypogonadism. One case reports phenotypically normal parents heterozygous for a GnRHR mutation who had a son with normal puberty and normal olfaction but with small (8-mL) testes and an abnormal semen analysis. Their daughter had primary amenorrhea and was infertile. LH pulse frequency was normal but with low amplitude pulsation.
Other reports describe GnRHR mutations causing hypogonadotropic hypogonadism that presents with complete gonadotropin deficiency. An example is a male patient seeking treatment for delayed puberty who presented with no secondary sexual characteristics, cryptorchid testes, low gonadotropins, and low testosterone. The patient did not respond to exogenous GnRH, but treatment with gonadotropins corrected testicular growth and descent, confirming a defect at the level of the GnRHR.
Recently, homozygous mutations in GNRH1, the genetic precursor to GnRH, have been shown to be a rare cause of normosmic IHH. The GNRH1 mutation is inherited in an autosomal recessive pattern. Administration of exogenous pulsatile GnRH restores the hypothalamic-pituitary-gonadal axis in these patients. [27]
DAX1 gene
Adrenal hypoplasia congenita arises from X-linked or autosomal recessive syndromes and presents in infancy with primary adrenal insufficiency. Treatable with steroids, it has resulted in affected adults developing hypogonadotropic hypogonadism. A pituitary origin for one group with hypogonadotropic hypogonadism has been suggested by the failed attempts in those patients to stimulate LH and FSH with pulsatile GnRH. A smaller group has had gonadotropin responses to GnRH therapy, characterizing a hypothalamic-versus-pituitary defect.
The DAX1 gene has been identified at Xp21 as the gene responsible for adrenal hypoplasia congenita. As with the KAL gene, there is a growing body of evidence that DAX mutations result in a wide phenotypic range. These data suggest that DAX1 mutations impair gonadotropin production via defects at the levels of both the pituitary and the hypothalamus. One suggested role for DAX1 is as a "brake" for normal male maturation, while also being necessary for normal adrenal and hypothalamic/pituitary development. DAX1 has been shown to block steroidogenesis in adrenal cells by transcriptional repression. Indeed, loss of function of this repressor may lead to a host of adrenal, hypothalamic, and pituitary abnormalities.
Additionally, steroidogenic factor 1 (SF-1), a nuclear hormone receptor for DAX1 (dosage-sensitive sex reversal, adrenal hypoplasia critical region, on chromosome X, gene 1), plays a regulatory role in adrenal development and development of the hypothalamic-pituitary-gonadal axis. [28] Specifically, SF-1 regulates expression of the p450 steroid hydroxylase genes in the gonads and the adrenal cortex, Mullerian Inhibitory Substance (MIS), the alpha subunit of the gonadotropins, and the beta subunit of LH.
Leptin and leptin receptor
Mutations in either leptin, a cytokine secreted from adipocytes that serves as a central satiety signal and a permissive signal to the reproductive system, or the leptin receptor lead to normosmic hypogonadotropic hypogonadism. Patients with this rare disorder fail to progress through puberty without exogenous leptin administration. The major associated phenotypic feature is obesity due to hyperphagia, which is also attenuated by leptin treatment. [29]
TAC3 and TACR3
Recently, analysis of single nucleotide polymorphisms (SNPs) among families with multiple members affected by IHH have identified autosomal recessive mutations in TAC3 and its receptor, TACR3, as another cause of IHH. [30] TAC3 encodes for neurokinin B, which is the ligand for the neurokinin-3 receptor (TACR3), Patients with mutations in TAC3 or TACR3 have isolated IHH without other phenotypic features, suggesting TAC3 and TACR3 function specifically to promote GnRH release. In fact, neurokinin B is found co-localized with kisspeptin and dynorphin in neurons of the arcuate nucleus of the hypothalamus. These neurons project to the median eminence and are closely opposed to GnRH neurons. Further, GnRH neurons have been shown to express TACR3.
Communication between GnRH neurons and neurons co-expressing kisspeptin, dynorphin, and neurokinin B has been proposed to represent the "GnRH pulse generator." [12] A study by Nagae et al provided direct evidence in rats that kisspeptin/neurokinin B/dynorphin A (KNDy) neurons in the arcuate nucleus of the hypothalamus generate GnRH pulses. [31]
NELF
Nasal epithelial LHRH factor (NELF) is involved in GnRH and olfactory neuronal development and has been implicated in rare cases of IHH. NELF co-localizes with GnRH in stem cells of the olfactory system. Heterozygous mutations have been identified in only 2 reported cases of IHH; thus, the role of NELF as a genetic cause of IHH has not been fully elucidated. [32]
Others
Advances in molecular genetics have lead to the discovery of several additional candidate genes for KS and IHH, and the future holds much more to be discovered in this area. These include SEMA3A, a semaphorin protein family member that is necessary for GnRH neuron development due to its role as a guidance cue for GnRH neuron migration. Lack of SEMA3A signaling in mice causes hypogonadal hypogonadism, and this mutation has been described in one case of human KS. [33] Missense mutations in WDR11, a gene involved in olfactory neuron development and human puberty, have also recently been described in patients with KS and IHH. [34]
Although most cases of IHH have been attributed to single gene defects, Pitteloud et al reported 2 families with this condition but with 2 different gene mutations. [22] With oligogenic mutations resulting in compound heterozygotes, synergistic effects of the mutated genes are hypothesized to result in hypothalamic hypogonadism. Since this initial finding by Pitteloud, several additional cases of oligogenic mutations have been identified in patients with KS and normosmic IHH. Mutations of PROKR2 + GPR54, PROKR2 + GnRHR, PROKR2 + KAL1, PROKR2 + FGFR1, PROKR2 + PROK2, FGFR1 + NELF, FGFR1 + GnRHR, and FGFR1 + FGF8 have been identified.
Interestingly, in addition, one patient normosmic IHH and 3 different mutations has been identified to date (PROKR2, GnRHR, and FGFR1). [35, 21] Furthermore, a study of a large cohort of patients suggests that oligogenicity is the norm in KS and IHH, rather than monogenicity. [36] With the advanced technology available for genetic analysis and with the identification of the human genome, scientists are constantly shedding new light on the complex genetic transmission of KS and IHH. This oligogenic model may explain the phenotypic variability observed within and across families with single gene defects.
Furthermore, cases of adult-onset and reversible IHH suggest that not only are genetic abnormalities involved in the pathogenesis of this disorder but that nongenetic factors may also contribute, such as hormonal and/or environmental factors. These have yet to be elucidated but research is ongoing.
An analysis of a cohort of 81 Greek isolated GnRH deficiency patients found the prevalence of normosmic idiopathic hypogonadotropic hypogonadism higher than Kallmann syndrome (67% to 33%) and putative causal genetic change was discovered in approximately 21% of the cohort. [37]
Epidemiology
United States statistics
The incidence of KS in the United States is 1 case per 10,000 men and 1 case per 50,000 women. The incidence of normosmic IHH is also rare and is estimated to be around 1 case in 70,000 to 1 case in 100,000 individuals.
International statistics
By examining military records, the incidence of KS has been estimated to be between 1 case per 86,000 in Sardinia and 1 case in 10,000 in France. [38]
Race-, sex-, and age-related demographics
Race
Race is not a factor in incidence.
Sex
In a referral population at Massachusetts General Hospital over a 20-year period, the male-to-female ratio was 3.9 to 1. [10]
A spectrum of GnRH deficiency, with various secretory patterns ranging from complete lack of LH pulsatility to diminished pulse amplitude similar to early puberty, occurs in both men and women, contributing to the clinical heterogeneity of the disorder. This suggests that multiple genetic determinants may control the expression of GnRH secretion.
Age
The disease comes to attention when the patient fails to begin puberty and does not develop secondary sexual characteristics.
Prognosis
Morbidity/mortality
These patients are not known to have an increased mortality rate; however, prolonged deficiency in gonadal hormones contributes to increased morbidity and may contribute to the aging process.
Patient Education
For excellent patient education resources, visit eMedicineHealth's Thyroid and Metabolism Center, Women's Health Center, and Pregnancy Center. Also, see eMedicineHealth's patient education articles Hypopituitary, Anatomy of the Endocrine System, Menopause, Amenorrhea, Birth Control Overview, and Birth Control Methods.
-
Human GPR54 receptor model. Mutations identified in patients with idiopathic hypogonadotropic hypogonadism are indicated.
-
KiSS-1 protein product model. Amino acids 1-19 are predicted to form a signal peptide. Proteolytic processing is predicted to produce kisspeptin-54, corresponding to amino acids 68-121. Shown is the C-terminal amidated decapeptide sequence, wherein biologic actively resides.






