Practice Essentials
Hürthle cell carcinoma of the thyroid gland is an unusual and relatively rare type of differentiated thyroid cancer. [1] Hürthle cell cancer accounts for only about 3-10% of all differentiated thyroid cancers; therefore, few institutions have extensive experience with Hürthle cell neoplasms. See the image below.
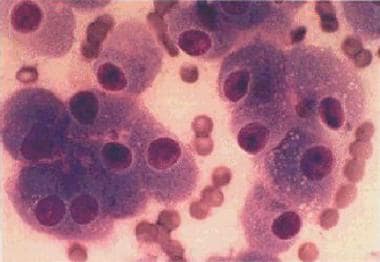 Hürthle cell carcinoma. A monomorphous cell population of Hürthle cells arranged in loosely cohesive clusters and single cells. The cells are polyhedral and have abundant granular cytoplasm with well-defined cell borders. The nuclei are enlarged and have a central prominent macronucleolus.
Hürthle cell carcinoma. A monomorphous cell population of Hürthle cells arranged in loosely cohesive clusters and single cells. The cells are polyhedral and have abundant granular cytoplasm with well-defined cell borders. The nuclei are enlarged and have a central prominent macronucleolus.
Hürthle cells are observed in both neoplastic and nonneoplastic conditions of the thyroid gland (eg, Hashimoto thyroiditis, nodular and toxic goiter). [2] Oncocytic cells in the thyroid are often called Hürthle cells, and oncocytic change is defined as cellular enlargement characterized by an abundant eosinophilic granular cytoplasm as a result of accumulation of altered mitochondria. This is a phenomenon of metaplasia that occurs in inflammatory disorders, such as thyroiditis, or other situations that result in cellular stress. The proliferation of oncocytes gives rise to hyperplastic and neoplastic nodules. [3] The cytologic features for Hürthle cell neoplasms are hypercellularity with a predominance of Hürthle cells (usually > 75%), few or no lymphocytes, and scanty or absent colloid.
Hürthle cells were first described by Askanasy in 1898, in patients with Graves disease; however, these cells were mistakenly named for the German physiologist Karl Hürthle, who actually described the interfollicular C cell. [4] Hürthle cells are large and polygonal in shape, with indistinct cell borders. They have a large pleomorphic hyperchromatic nucleus, a prominent nucleolus, and intensely pink, fine, granular cytoplasm with hematoxylin-eosin staining.
Hürthle cells are also found in other tissues, such as the salivary gland, parathyroid gland, esophagus, pharynx, larynx, trachea, kidney, pituitary, and liver. Controversy exists about the origin of Hürthle cells, which generally are thought to derive from the follicular epithelium.
Although Hürthle cell carcinoma was previously considered a variant of follicular cell neoplasms, which are generally less aggressive, the 2017 World Health Organization (WHO) classification of endocrine tumors reclassified it as a distinct entity. The WHO defines Hürthle cell carcinoma morphology as minimally invasive capsular invasion and widely invasive vascular invasion of > 4 blood vessels. Molecular markers include RAS, EIF1AX, PTEN, TP53, CNA, mtDNA. [5] The 2022 WHO classification of endocrine tumors has replaced the term Hürthle cell carcinoma with oncocytic carcinoma. [6]
A benign neoplasm cannot be distinguished from a malignant neoplasm on the basis of cytologic analysis of a fine-needle aspiration (FNA) biopsy specimen. Features such as pleomorphism, anaplasia, hyperchromatism, and atypia are also observed in benign follicular adenomas; therefore, definitive differentiation of Hürthle cell carcinoma from Hürthle-cell adenoma is based on vascular invasion and/or capsular invasion, as well as on permanent histologic sections or extrathyroidal tumor spread and lymph node and systemic metastases.
In the literature, the incidence of malignancy in Hürthle-cell neoplasms is variable, ranging from 13-67%. Overall, only about 33% of Hürthle cell tumors demonstrate signs of that invasive growth that indicates malignancy and the possibility of metastasis. On balance, Hürthle cell tumors may be considered to be more likely to metastasize than follicular tumors. The likelihood of nodal metastases is greater in Hürthle cell tumors than in follicular tumors; it is, however, not as great as with papillary tumors.
Permissive histologic interpretation may result in the designation of some non-neoplastic Hürthle cell lesions as malignant tumors. Obviously, this factor has a major impact in interpreting the natural history of this disease and adds to the controversy about the aggressiveness of Hürthle cell carcinoma. This leads to reported overall mortality rates ranging from 9-28%.
Tumor size is an important feature for biological behavior. A 1988 study found that a Hürthle tumor that is 4 cm or larger has an 80% chance of histologic evidence of malignancy. [7] In another study by Pisanu et al, [8] in a series of 23 patients, the mean tumor size was significantly greater for carcinomas than adenomas (3.1 cm vs 1.9 cm).
In a study of 56 patients with Hürthle cell cancer treated at Memorial Sloan-Kettering Cancer center, recurrence was a significant predictor of tumor-related mortality, and extent of invasion was the most significant predictor of outcome. [9] A study of 111 patients with Hürthle cell carcinoma found that recurrence-free survival rates were 100% in patients without vascular invasion, 95% in those with focal vascular invasion, and 77% in those with extensive vascular invasion. [10]
In a study of patients with Hürthle cell thyroid neoplasm who underwent surgery for suspected carcinoma, the histopathologic diagnosis was carcinoma in 71 of 279 patients. Predictive factors for carcinoma were age older than 65 years and thyroglobulin concentration over 1000 ng/mL. [11]
Hürthle cell cancer reportedly behaves in a more aggressive fashion than other well-differentiated thyroid cancers, with a tendency to higher frequency of metastasis and a lower survival rate. This is truer for the lesions that are clearly demonstrated to be malignant and in patients who are considered to be at high risk based on such factors as age, tumor size, invasiveness, and the presence of metastasis. Widely invasive tumors behave more aggressively. Recurrent Hürthle cell carcinomas are considered to be incurable.
Hürthle cell cancer has the highest incidence of metastasis among the differentiated thyroid cancers. Metastatic disease is reported at the time of initial diagnosis in 10-20% of patients and in 34% of the patients overall. Metastasis usually occurs hematogenously, but lymph node metastasis is also not uncommon and typically involves the regional lymph nodes. Some studies suggest that lymph node metastases at initial diagnosis may not be an unfavorable prognostic factor. [12] The lungs, bones, and central nervous system are the most prevalent sites of metastases.
Pathophysiology
No widely accepted paradigm exists for the pathogenesis of follicular and Hürthle cell cancer of the thyroid. Some evidence suggests that a multistep adenoma-to-carcinoma pathway may be involved; however, this concept is not universally accepted. Many of the cells probably develop from preexisting adenomas, but a follicular carcinoma in situ is not recognized pathologically.
Progressive transformation through somatic mutations of genes that are important in growth control are involved in follicular thyroid cancer formation. Low iodide intake is a key environmental factor determining the relative incidence of follicular and papillary cancers. Most follicular adenomas and all follicular carcinomas are thought to have monoclonal origin.
Oncogene activation, particularly by mutation or translocation of the ras oncogene, is common in both follicular adenomas and follicular thyroid carcinomas (around 40%), supporting a role in early tumorigenesis. Such ras oncogene mutations are not specific for follicular tumors and also occur in papillary thyroid cancer (PTC). The RAS oncogene is frequently involved in the pathogenesis of Hürthle cell tumors.
In papillary thyroid cancers and in many Hürthle cell tumors, RET rearrangements are found; these are not found in follicular tumors. Local spread may be found in RET-positive cases; RET-negative cases, as in follicular cancer cases, are more likely to spread through the bloodstream to distant metastatic sites.
An association also was found between overexpression of the p53 gene product and a subset of Hürthle cell carcinomas. Reduced immunoexpression of E-cadherin exists, with a trend to a diffuse cytoplasmic pattern, both in benign and malignant Hürthle cell tumors and in papillary, poorly differentiated, and undifferentiated thyroid carcinomas. Isolated studies indicate overexpression of the N-myc oncogene, tumor growth factor (TGF)-alpha, TGF-beta, insulinlike growth factor (IGF)-1, and somatostatin receptor in Hürthle cell carcinomas.
Cytogenetic abnormalities and evidence of genetic loss are more common in follicular thyroid cancer than in papillary thyroid cancer. These abnormalities occur in follicular adenomas, suggesting that cell cycle control, mitotic spindle formation, DNA repair, or more than one of these mechanisms may be impaired in these neoplasms, possibly at an earlier stage.
Activating mutations of genes encoding the thyrotropin receptor and the alpha subunit of the stimulatory G protein are also reported in some follicular carcinomas. These losses are associated particularly with chromosomes 3, 10, 11, and 17. The deletions and/or rearrangements involving the p (short) arm of chromosome 3 are the most common. Loss of a tumor suppressor on chromosome arm 3p has been postulated to be specific for follicular thyroid cancer and may be involved in adenoma-to-carcinoma progression.
Restriction fragment length polymorphism (RFLP) analysis demonstrates that unbalanced losses of genetic material are relatively common in Hürthle cell neoplasms. Loss of heterozygosity from the q (long) arm of chromosome 10 is also detected in oncocytic tumors.
Evidence suggests that some Hürthle cell adenomas and carcinomas can express an RET/PTC gene arrangement. Because this gene arrangement is more specific to papillary thyroid carcinoma, another subclassification of Hürthle cell neoplasms has been proposed—namely the papillary variant of Hürthle cell cancer (ie, Hürthle cell papillary thyroid carcinoma)—in addition to Hürthle cell cancer and adenoma. Clinically, tumors in this group tend to behave like papillary thyroid carcinoma; however, they are more indolent, with a propensity for lymph node metastasis rather than hematogenous spread. Maxwell et al reported that the Hürthle cell tumors with RET/PTC-positive gene arrangement have a higher incidence of regional metastatic disease, and more aggressive treatment has been recommended. [13]
As reported by Asa, many Hürthle cell tumors, whether benign or malignant, show papillary change. This is a pseudopapillary phenomenon because Hürthle cell tumors have only scant stroma and may fall apart during manipulation, fixation, and processing. True oxyphilic, or Hürthle cell, papillary carcinoma has been reported to comprise 1%-11% of all papillary carcinomas. These tumors have a papillary architecture but are composed predominantly, or entirely, of Hürthle cells. [3]
Mitochondrion-related alterations, such as mutations in mitochondrial DNA, are also described in Hürthle cell tumors. Defects of cytochrome c oxidase and the deletion of mitochondrial DNA occur frequently in Hürthle cell tumors and in Hürthle cells of Hashimoto thyroiditis. In one study, almost all Hürthle cells displayed a common deletion, somatic mitochondrial point mutations, or both. [14] Activating gene mutations encoding the thyrotropin receptor and the alpha subunit of the stimulatory G protein are also reported in some follicular carcinomas.
DNA content profiles after flow cytometry are commonly abnormal. Hürthle cell neoplasms, including histologically benign tumors, are often aneuploid. This finding parallels with nuclear atypia and anisocytosis. The demonstration of aneuploidy may be a marker for a particularly aggressive clinical behavior compared with euploid tumors. In an Italian study, p27 and cyclin D3 proteins were found to be overexpressed in Hürthle cell carcinoma cell lines and clinical samples of thyroid cancer. [15] The accumulation of p27 was found to be associated to the overexpression of cyclin D3 in Hürthle cell carcinoma of the thyroid.
A study analyzing genomic dissection of Hurthle cell carcinoma indicated that Hurthle cell carcinoma could be a unique type of malignancy. In this study, unsupervised hierarchical clustering of gene expression showed 3 groups of Hurthle cell tumors: Hurthle cell adenomas, minimally invasive Hurthle cell carcinoma, and widely invasive Hurthle cell carcinoma. These are clustered separately, with a marked difference between widely invasive Hurthle cell carcinoma and Hurthle cell adenoma. Molecular pathways that differentiate Hurthle cell adenomas from widely invasive Hurthle cell carcinomas included the PIK3CA-Akt-mTOR and Wnt/β -catenin pathways, potentially providing a rationale for new targets for the treatment of this type of thyroid carcinoma. [16]
Etiology
Etiologic factors in Hürthle cell carcinomas include the following:
-
Radiation to the neck
-
Iodide deficiency
-
Overexpression of the p53noncogene
-
Activating mutations of genes encoding the thyrotropin receptor and the alpha subunit of the stimulatory of G protein are reported in some follicular carcinomas
-
Somatic mutations of genes important in growth control
-
Oncogene activation, particularly by mutation or translocation of the RAS oncogene
-
Mitochondrion-related alterations (eg, mutations in mitochondrial DNA) also are described.
A study by Maximo et al linked somatic and germline mutation in GRIM-19 (a dual-function gene involved in mitochondrial metabolism and cell death) to Hürthle cell tumors of the thyroid. This is the first nuclear gene mutation described for a subgroup of Hürthle cell carcinomas. [17]
Epidemiology
Frequency
United States
The American Cancer Society (ACS) estimates that 43,720 new thyroid cancers will be diagnosed in 2023, 31,180 in women and 12, 540 in men; the ACS estimates that 2120 deaths from thyroid cancer will occur, 1150 in women and 970 in men. In women, thyroid cancer is the seventh most common cancer, accounting for approximately 3% of all new cases. [18] Hürthle cell carcinoma is rare and represents only about 3% of all thyroid cancer diagnoses. [19]
International
The European Network of Cancer Registries report that the incidence of thyroid cancer varies from country to country. Lithuania reported the highest incidence per 100,000 person-years (15.5) followed by Italy (13.5), Austria (12.4), Croatia (11.4), and Luxembourg (11.1). [20]
Race- and age-related demographics.
All races appear to be affected equally. The typical age range of patients presenting with this condition is 20-85 years. The mean age is usually 50-60 years, approximately 10 years older than the age associated with other types of differentiated thyroid cancers.
Prognosis
Hürthle cell carcinomas behave in a more aggressive fashion than other well-differentiated thyroid cancers, as evidenced by a higher incidence of metastasis and a lower survival rate. Hürthle cell carcinomas produce thyroglobulin. In addition, most Hürthle cell carcinomas have decreased avidity for iodine-131; therefore, treatment with radioactive iodide has limited efficacy.
In some series, nuclear aneuploidy is present in as many as 90% of patients with Hürthle cell carcinoma; in some studies, this condition is shown to be associated with an adverse prognosis.
Ghossein et al at Memorial Sloan Kettering Cancer center reported that in encapsulated ("minimally invasive") Hürthle cell carcinomas, the extent of vascular invasion strongly correlated with recurrence. Presence of mitosis and a solid/trabecular tumor growth pattern also correlated with higher risk of recurrence. [21]
Mortality rates vary in different series, based on the staging systems used, which consider the patient's age, tumor size, extrathyroidal tumor spread, pathologic classification of the neoplasm (Hürthle cell carcinoma versus adenoma), and the therapeutic approach.
Overall survival rates reportedly are similar or worse in patients with Hürthle cell carcinoma compared with rates for persons with follicular carcinoma. In a case series of Hürthle cell carcinoma, mortality rates at 5, 10, and 20 years were 8%, 18%, and 33%, respectively. Two other case series confirmed a 20-year cause-specific mortality rate of 20-35%. One study showed that when distant metastases were present, the 5-year mortality rate was 65%. [22] Another study involving 33 patients showed that disease-free survival was 65% in 5 years and 40.5% in 10 years. [23]
In a study of 108 patients with metastatic Hürthle cell thyroid carcinoma, Besic et al reported that sites of metastasis, in decreasing order of frequency, were lung, bone, mediastinum, kidney, and liver. Overall 10-year disease-specific survival was 60%. Median disease-specific survival after the diagnosis of metastatic disease was 72 months for patients with pulmonary metastases and 138 months for patients with metastases at other sites. [24]
In a retrospective review of all patients treated with Hürthle cell carcinoma at their institution between 1946 and 2003 (62 patients in all), Mills et al found that independent predictors of disease-free survival were lymph node status (P = 0.008), presence of metastases at diagnosis (P = 0.005), and tumor stage (P = 0.009). These authors suggest that radical surgery may improve outcome; on multivariate analysis, extent of surgery (P < 0.001) was the only independent factor that affected cause-specific survival. [25]
In a large retrospective study that analyzed the Surveillance, Epidemiology, and End Results (SEER) database from 1988-2009, 3311 patients with Hürthle cell cancer were identified and compared with 59,585 patients with other types of differentiated thyroid cancer. Overall disease-specific survival rates were lower for patients with Hürthle cell cancer (P < 0.001), indicating that Hürthle cell cancer has more aggressive behavior and compromises survival more than other types of differentiated thyroid cancer. [26]
In a study of 239 patients with Hürthle cell cancer treated at a single institution from 1995 to 2014, Oluic et al reported 5-, 10-, and 20-year cancer-specific survival rates of 94.6%, 92.5%, and 87.4%, respectively. Involvement of both thyroid lobes and the need for reoperation due to local relapse were unfavorable independent prognostic factors, while total thyroidectomy as the primary procedure was a favorable predictive factor for cancer-specific survival. [27]
Patient Education
The need for life-long levothyroxine treatment should be explained to all patients. Radiation precautions should be explained clearly and in detail to patients who will be receiving radioactive iodine treatment. Women of childbearing age should be advised not to become pregnant for at least 1 year after treatment with131I .
For patient education information, see the Thyroid Cancer Directory.
-
Tumor, lymph node, metastases (TNM) staging system for papillary and follicular thyroid carcinoma.
-
Hürthle cell carcinoma. A monomorphous cell population of Hürthle cells arranged in loosely cohesive clusters and single cells. The cells are polyhedral and have abundant granular cytoplasm with well-defined cell borders. The nuclei are enlarged and have a central prominent macronucleolus.






