Overview
Amyotrophic lateral sclerosis (ALS), also known as Charcot's disease and Lou Gehrig's disease, is a disease of unknown cause characterized by slowly progressive degeneration of upper motor neurons (UMNs) and lower motor neurons (LMNs). [1]
The UMN findings include hyperreflexia and spasticity. They result from degeneration of the lateral corticospinal tracts in the spinal cord. The LMN findings include weakness, atrophy, and fasciculations. They are a direct consequence of muscle denervation via destruction of the anterior horn cell. ALS is eventually fatal because of respiratory muscle weakness. Aspiration pneumonia and medical complications of immobility contribute to morbidity. [2] Although ALS is incurable, there are treatments that can prolong meaningful quality of life; therefore, diagnosis is important to patients and families.
Also see the following:
Diagnosis
Emergency physicians should be familiar with amyotrophic lateral sclerosis (ALS) and should consider the diagnosis in patients with motor symptoms and signs of hyperreflexia.
The diagnosis of ALS is primarily clinical. Electrodiagnostic testing contributes to the diagnostic accuracy by objectively looking for lower motor neuron (LMN) involvement.
ALS can be differentiated from stroke or trauma due to the subacute or chronic progression of symptoms. When focal limb weakness occurs, ALS is differentiated from a root or peripheral nerve lesion by the lack of pain or sensory symptoms and possibly the presence of signs of hyperreflexia. While ALS is a slowly progressive disease, a precipitous event may occur to bring the patient to the emergency department (ED), such as an infection or respiratory compromise.
Bulbar symptoms
The patient's family first notices slurring of words or choking during a meal. An aspiration event or acute respiratory symptoms of air hunger occur. Patients may present to the ED with either an aspiration event or neuromuscular respiratory failure with CO2 retention. Often, they will be very anxious at night due to hypercarbia.
Extremity weakness
The patient notices wrist drop interfering with his or her work performance. Alternatively, the patient may find reduced finger dexterity, cramping, stiffness, and weakness or wasting of intrinsic hand muscles. The patient may develop painless foot drop resulting in a fall or sprain. The fall that may result can be the presenting feature in the ED.
Fasciculations may present early on in the disease, particularly in the tongue.
Emergency Department Care
Patients initially are cared for symptomatically, and emotional support should be available to the patient and family. Discussion of a new diagnosis of ALS in the ED is inappropriate before further evaluation occurs.
If a living will or a declaration for a natural death is in place, keep patients comfortable and do not intubate if this is consistent with the patient's wishes.
Complications, such as infections, deep vein thrombosis, or respiratory insufficiency, should be managed accordingly.
Further information on the diagnosis of and treatment for ALS is available from the European Federation of Neurological Societies. [3]
Consultations
Refer patients to a neurologist, preferably a neuromuscular specialist experienced in the diagnosis and treatment of neuromuscular diseases.
Refer to a pulmonologist if respiratory failure is imminent and patient wishes allow.
Inpatient Care
Physical and occupational therapy and speech pathology consultations assist the patient with amyotrophic lateral sclerosis (ALS) in maintaining strength, daily living activities, and communication skills (as demonstrated in the image below).
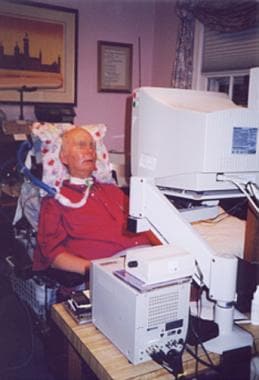 Patient with amyotrophic lateral sclerosis (ALS) using eye-gaze computer device to conduct business and communicate.
Patient with amyotrophic lateral sclerosis (ALS) using eye-gaze computer device to conduct business and communicate.
When forced vital capacity (FVC) reaches about 50% of predicted levels, supplemental respiratory support may be needed. (The first symptom may be disturbed sleep.) Use of noninvasive positive pressure ventilation improves quality of life; further respiratory deterioration may necessitate tracheostomy and ventilator support.
Patients with progressive weight loss, symptomatic dysphagia, or aspiration should be considered for percutaneous gastrostomy placement. Risk of percutaneous endoscopic gastrostomy (PEG) placement increases with declining respiratory function. PEG is best placed before FVC falls to less than 50% of predicted levels.
Advance directives may be addressed by the patient's neurologist and be discussed at regular intervals. Hospice referral may be appropriate to aid in care during the terminal phase of the disease.
Medications
Treatment of pneumonia, which is a complication of amyotrophic lateral sclerosis (ALS), [2] initially involves the empiric use of a relatively broad-spectrum antibiotic or antibiotics effective against probable pathogens, after appropriate cultures and specimens for laboratory evaluation have been obtained. These medications may include cephalosporins, fluoroquinolones, vancomycin, penicillins, and aminoglycosides.
Amyotrophic lateral sclerosis agent
Two disease-specific medications are approved by the US Food and Drug Administration (FDA) for use in patients with ALS: riluzole and edaravone.These should be prescribed only by a physician familiar with inclusion/exclusion criteria and who will be monitoring patient. They should not be prescribed in the ED acutely. [1, 4]
For patients who develop fatigue while taking riluzole, withholding the drug may be considered.
Antispasticity agents
Treatment of spasticity is not specific for amyotrophic lateral sclerosis (ALS). Oral antispasticity agents that may be used include baclofen, benzodiazepines, tizanidine, gabapentin, or dantrolene. [12]
Cholinergic agents
These can be used in patients with ALS who may have secondarily impaired neuromuscular junction transmission.
Antisialorrhea agents
Sialorrhea, or drooling, is embarrassing and is associated with aspiration pneumonia. The prevalence for patients with ALS is 50%. Anticholinergic medications are generally tried first to reduce sialorrhea, although effectiveness is unproven. [5]
In October 2009, the American Academy of Neurology (AAN) published a 2-part, evidence-based practice parameter update about the care of the patient with ALS. [6, 7] The practice parameter recommended that botulinum toxin B (rimabotulinumtoxinB [Myobloc]) be considered for the treatment of refractory sialorrhea (Level B evidence) and that low-dose radiation therapy to the salivary glands be considered as well (Level C evidence). [8]
Pseudobulbar affect
Pseudobulbar affect, excessive laughing or crying or involuntary emotional expression, affects 20-50% of patients with ALS. [9] combined dextromethorphan and quinidine has been administered for this condition [10] . This drug combination was approved by the FDA in October 2010.
Questions & Answers
Overview
What is amyotrophic lateral sclerosis (ALS)?
How is amyotrophic lateral sclerosis (ALS) diagnosed?
How is amyotrophic lateral sclerosis (ALS) differentiated from stroke or trauma?
What are the bulbar symptoms of amyotrophic lateral sclerosis (ALS)?
What are the muscular symptoms of amyotrophic lateral sclerosis (ALS)?
Which specialist consultations are beneficial to patients with amyotrophic lateral sclerosis (ALS)?
What is included in the inpatient care of amyotrophic lateral sclerosis (ALS)?
What is the role of medications in the treatment of amyotrophic lateral sclerosis (ALS)?
What is the role of riluzole in the treatment of amyotrophic lateral sclerosis (ALS)?
What is the role of cholinergic medications in the treatment of amyotrophic lateral sclerosis (ALS)?
How is pseudobulbar affect treated in patients with amyotrophic lateral sclerosis (ALS)?
-
Patient with amyotrophic lateral sclerosis (ALS) using eye-gaze computer device to conduct business and communicate.







