Background
Niemann-Pick disease (NPD) comprises an autosomal recessively inherited (see diagram at end of section) group of congenital lipidoses in which sphingolipids accumulate in cells, especially reticuloendothelial cells, throughout the body.
The following six types of Niemann-Pick disease have been described:
-
Type A - Acute neuronopathic form
-
Type B - Visceral form
-
Type C - Chronic neuronopathic form
-
Type D - Nova Scotia variant
-
Type E - Adult form
-
Type F - Sea-blue histiocyte disease
Other variants include an acute form with hydrops; an early form with neonatal hepatitis; and a more slowly evolving, chronic form with progressive neurologic deterioration that extends well into adulthood.
Niemann and Pick, and later Crocker and Farber, defined Niemann-Pick disease on the basis of its clinical and pathologic features in the beginning of the 20th century. The Niemann-Pick group of diseases can be subclassified into two categories: (1) those with a primary deficiency in acid sphingomyelinase (ASM) activity (ie, types A and B) and (2) those with defective intracellular processing and transporting of low-density lipoprotein (LDL)–derived cholesterol (ie, type C).
The disease is clinically characterized by progressive degeneration of the central nervous system with visceral accumulation of cholesterol and sphingomyelin. The clinical phenotype is extremely variable, ranging from an acute neonatal form, with mainly liver involvement and rapid neurologic deterioration, to an adult late-onset form, with slowly progressive ataxia and a movement disorder. The late infantile and juvenile forms are considered to be the most common classic presentations, with the insidious onset of ataxia, vertical supranuclear gaze palsy, and cognitive impairment in as many as 80% of patients. Foam-cell infiltration and visceromegaly are common features in all forms, but neurologic involvement occurs only in types A and C and not in type B.
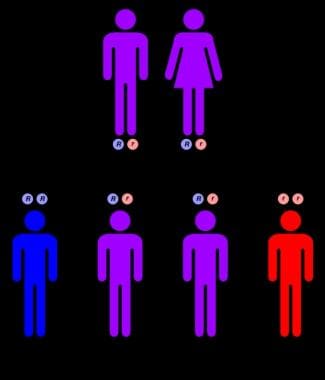 Autosomal recessive inheritance pattern.
Autosomal recessive inheritance pattern.
The pediatrics article Niemann-Pick Disease may be of interest, as may Lysosomal Storage Disease and Lipid Storage Diseases.
Pathophysiology
The sphingomyelin that accumulates in the lysosomes of Niemann-Pick disease–affected cells is thought to arise from the degradation of the cells and their organelles because it is a major component of all mammalian cell membranes. In Niemann-Pick disease type C, the main lipid that accumulates in patients' cells is not sphingomyelin but cholesterol; however, sphingomyelin metabolism and cholesterol metabolism are closely related.
Sphingomyelinase is an acidic lysosomal hydrolase that catalyses the cleavage of sphingomyelin to phosphoryl choline and ceramide. In patients with Niemann-Pick disease, its activity is deficient in all lysosome-containing tissues. In patients with Niemann-Pick disease type A, the infantile form, sphingomyelinase activity is 0.7% of that of healthy individuals, whereas in patients with adult-onset neuronopathic or nonneuronopathic disease, the activity is 0-19% of that of healthy individuals.
This enzyme defect explains the massive deposition of sphingomyelin in tissues of the reticuloendothelial systems. In patients with the group A variant, sphingomyelin and other lipids are stored in the brain in increased amounts, a finding consistent with the neuronopathic features, whereas in patients with the group B form, the nervous tissue does not appear to store sphingomyelin.
In both healthy individuals and patients with Niemann-Pick disease types A and B, fibroblasts synthesize sphingomyelinase polypeptides with the same molecular mass of 110 kd, in the same amount. During further processing, the 110-kd polypeptide is reduced to a molecular weight of 84 kd. The deficiency of sphingomyelinase is due to intragenic defects.
Findings from experiments conducted so far suggest that the specific defects could be small inframe deletions, inframe additions, or point mutations. The differences in the clinical courses of types A and B suggest that the mutations are different. Sphingomyelinase follows the same intracellular targeting and posttranslational processes as most of the lysosomal hydrolases. However, unlike any other enzyme, the polypeptide exists in 2 forms of different sizes. Each polypeptide is differentially distributed in the tissues. In tissues, such as the brain, the smaller polypeptide (80 kd) is found, whereas the kidneys contain both polypeptides (110 and 80 kd). No precise explanation exists for the occurrence of one form of the polypeptide in some tissues and the presence of both forms in tissues, such as the kidneys.
The discovery of the NPC1 gene by Carstea et al [1] in 1997 has stimulated much research in Niemann-Pick disease, the NPC1 gene product, sterol trafficking, and gene therapy in animal models. The NPC1 gene was cloned after it was mapped to the long arm of chromosome 18 by using linkage and positional cloning techniques. The NPC1 complementary DNA (cDNA) sequence suggests a protein of 1,278 amino acids with an estimated molecular mass of 142 kd. Topological analysis of the NPC-1 protein has revealed the existence of 13 transmembrane domains, 7 luminal loops, 6 cytoplasmic loops, and a cytoplasmic tail with a signal sequence for targeting to the endoplasmic reticulum and a dileucine motif that directs the NPC-1 protein to lysosomes.
Of particular importance is the presence of a sterol-sensing region in the NPC1 sequence that has extensive homology to other mediators of cholesterol homeostasis, including 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase and sterol regulatory element–binding protein cleavage-activation protein. The region also has homology to the human PATCHED gene, which serves as the transmembrane receptor for sonic hedgehog morphogen. Defective sonic hedgehog morphogen in humans can lead to basal cell nevoid syndrome and holoprosencephaly.
To date, more than 100 NPC1 mutations, mostly missense mutations, have been identified throughout the gene, with no apparent hot spots. Most patients with NPC1 mutations have compound heterozygosity with unique mutations. The 2 known exceptions are a common I1061T mutation that is found in the United Kingdom; in France; and in the upper Rio Grande Valley in the southwestern United States, where it is present in the Hispanic population. The G992W mutation is found in the Acadian population of Nova Scotia.
NPC1 mutations are responsible for the disease in approximately 95% of patients. [2, 3] The NPC1 gene was evaluated in 5 Taiwanese/Chinese patients with NPC in the Republic of China. Six novel NPC1 mutations (N968S, G1015V, G1034R, V1212L, S738Stop, and I635fs) were identified, 3 of which were missense mutations located in the cysteine-rich domain.
The function of the NPC-1 protein is of great interest because it could enhance the understanding of the cholesterol exchange among the various subcellular compartments. The accumulation of unesterified cholesterol in NPC-1 lysosomes implies that NPC-1 is involved in the transport of free cholesterol from this organelle.
Three putative functions can be assigned to NPC-1, as outlined below.
-
NPC-1 may function as a cholesterol transporter by directly collecting cholesterol from the membranes of the endosomal-lysosomal system and transporting the lipid to the trans-Golgi network (TGN). Such a function could explain the transient interaction observed between the NPC-1 protein and lysosomes and the TGN. This function could also explain the observation that mutated NPC-1 protein localizes to the membranes of cholesterol-filled organelles but that it is unable to effect cholesterol mobilization.
-
NPC-1 may act as a docking and/or fusion protein that allows cholesterol-filled vesicles to dock and fuse with recycling endosomes for subsequent delivery to the TGN.
-
NPC-1 may act as a pump that drives the movement of cholesterol and possibly other lipids away from the endosome and to the TGN. However, NPC-1 lacks an ATP-binding cassette, which is typical in cellular molecular pumps.
In fibroblasts, the NPC1 gene product is localized to vesicles that test positive for lysosome-associated membrane protein-2 (LAMP2) and negative for the mannose 6-phosphate receptor. These vesicles transiently interact with cholesterol-laden lysosomes to facilitate sterol relocation. The cargo transported by means of this interaction is not limited to sterol but probably also involves glycolipids, which accounts for accumulations of glycolipids, such as GM2-ganglioside, in Niemann-Pick disease type C neurons and fibroblasts.
Sphingomyelin hydrolysis is a key component of a signal transduction pathway involved in cell proliferation, differentiation, and programmed cell death. A number of extracellular agents, including inflammatory cytokines, hormones, growth factors, nitric oxide, and other stressors, trigger the release of ceramide, which acts as an intracellular second messenger to regulate various cellular activities.
To further examine the role of ASM in cell signaling and apoptosis, investigators are using ASM-deficient cells obtained from patients with Niemann-Pick disease type A and cells obtained from ASM-deficient mice. In many instances, fully normal responses were observed, but, in others, the responses differed depending on whether Niemann-Pick disease cells or healthy cells were being tested. Correction of the enzyme deficiency by means of transfection with a plasmid encoding ASM could, in certain circumstances, correct the defect in signaling.
Bone marrow transplantation into newborn ASM knockout mice, performed by using donor cells from healthy animals in the colony, improves survival, delays the onset of ataxia, results in less lipid accumulation, and improves the histologic appearance of the brain and the visceral organs. Naturally occurring murine and feline models of Niemann-Pick disease type C that are clinically, biochemically, and morphologically equivalent to human Niemann-Pick disease type C have been characterized. Studies of apolipoprotein D metabolism in mice with Niemann-Pick disease type C show abnormalities in the apolipoprotein D gene and in protein expression. Plasma levels were increased 6-fold, and higher levels were also found in Niemann-Pick disease type C brain astrocytes and cultured astrocytes.
Because apolipoprotein D is important in cellular cholesterol transport for the synthesis, the assembly, and the maintenance of myelin, its sequestration could reflect the reduced myelin turnover and the deficiency in myelin, which are characteristic of Niemann-Pick disease type C.
One well-known NPC1 gene mutation causes a unique phenotype limited to descendants of a single Acadian ancestor in Nova Scotia, Canada. [4]
Etiology
See Pathophysiology.
An interesting parallel also exists between the up-regulation of apolipoprotein D in mice with Niemann-Pick disease type C and its enhanced expression in oligodendroglia in Alzheimer disease. Neurofibrillary tangles are a common neuropathologic feature of the two disorders; this finding suggests a relationship between apolipoprotein D and neurofibrillary tangles. Thus, the expression of apolipoprotein D appears to be coordinately impaired in Niemann-Pick disease type C as part of a generalized defect in cellular cholesterol trafficking.
Epidemiology
US frequency
Niemann-Pick disease type A occurs most frequently, and it accounts for about 85% of all cases of the disease. Niemann-Pick disease type C affects an estimated 500 children in the United States.
Race
One in 75 Ashkenazi Jews is a carrier.
Sex
Males and females are equally affected.
Age
Niemann-Pick disease affects infants, children, and adults. Niemann-Pick disease type A begins in the individual's first few months of life. Niemann-Pick disease type B has a more variable course, with the first symptoms occurring in early childhood. Many persons with Niemann-Pick disease type B survive into adulthood. Persons with Niemann-Pick disease type C have normal development for their first 2 years or more of life.
Prognosis
Patients with Niemann-Pick disease type A die in infancy. Patients with Niemann-Pick disease type B may live a comparatively long time, but many require supplemental oxygen because of lung involvement. The life expectancies of patients with Niemann-Pick disease types C and D are variable. Some patients die in childhood, whereas others who appear to be less drastically affected live into adulthood. The prognosis of Niemann-Pick disease type C correlates with the neurologic findings and age at onset; those with an early onset progress faster. [5]
Patient Education
Genetic counseling may be helpful. Carrier detection testing for all families is not yet reliable. The mutations in Niemann-Pick disease types A and B have been extensively studied, particularly in the Ashkenazi Jewish population, and DNA tests for these forms of Niemann-Pick disease are available. Antenatal diagnosis (ie, diagnosis in the fetus) of Niemann-Pick disease is possible in a limited number of centers. Carrier detection is possible in families only after their specific mutation is identified.
Niemann-Pick disease is inherited in an autosomal recessive manner. A couple who has a child with Niemann-Pick disease is at a 25% risk with each pregnancy of having another child who is affected; prenatal testing is available for such pregnancies. The phenotype (eg, age of onset, severity of symptoms) is usually consistent in families. Unaffected siblings of the proband have a 66% risk of being carriers of the gene for Niemann-Pick disease.
Biochemical testing can be used when the proband has a classic biochemical phenotype but not when the proband has a variant biochemical phenotype.
Molecular genetic testing can be used when the proband has mutations in the NPC1 gene that have been identified.
For patient education resources, see the Cholesterol Center and Statins Center, as well as High Cholesterol, Cholesterol FAQs, and Atorvastatin (Lipitor).
-
Autosomal recessive inheritance pattern.






