Practice Essentials
In 1929, Vohwinkel first described this syndrome in a 24-year-old woman who, since age 2 years, had a diffuse honeycombed palmar and plantar keratosis, in addition to distal interphalangeal creases. The constrictions ultimately led to autoamputation. The daughter of this patient experienced similar clinical lesions.
Pseudoainhum is the autoamputation of any digit secondary to keratodermas and other causes. In contrast, ainhum is the posttraumatic or postinfectious development of constricting bands of the digits resulting in autoamputation. Several categories can be distinguished, as follows:
-
Ainhum (dactylolysis spontanea): Ainhum is spontaneous autoamputation of the fifth toe, predominantly affecting blacks in tropical climates. This form most likely is posttraumatic or postinfectious in nature and is uncommon in the United States.
-
Congenital annular constricting bands
-
Autoamputation of traumatic origin, including self-mutilation and mechanical factors, frostbite, and burns
Signs and symptoms
The classic triad is as follows:
-
Diffuse palmoplantar keratoderma in a honeycombed pattern
-
Constricting bands of the digits with autoamputation (pseudoainhum)
-
Starfish-shaped hyperkeratotic plaques on the dorsum of the hands and feet, elbows, and knees (In addition, linear hyperkeratoses may be observed on the elbows and knees.)
Note the images below:
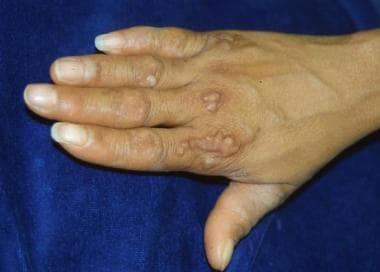
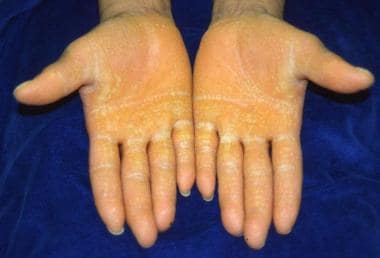
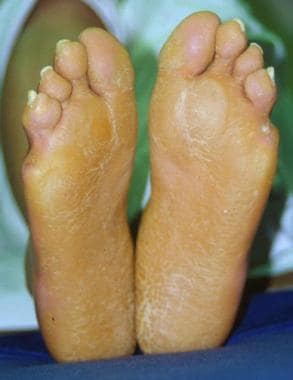
Rare associated findings include scarring alopecia and nonprogressive sensorineural hearing loss at high frequencies (classic variant).
A case report of two siblings described a probable new variant of Vohwinkel syndrome with congenital hypotrichosis. [1] Cases with intellectual disability [2] and an ichthyosiform presentation [3, 4] have also been described.
The classic skin lesions develop at an early age. Explore the family history for abnormalities suggestive of Vohwinkel syndrome.
Complications
Complications can include loss of digit(s) with functional impairment of the limb and hearing loss.
In two documented cases, squamous cell carcinoma has arisen secondary to Vohwinkel syndrome. [5]
Diagnostics
No invasive diagnostic procedures are routinely indicated.
Obtain radiographs of the hands and feet to detect abnormalities of the underlying bones. Use craniofacial imaging studies if clinical findings justify them.
Audiometry is suggested to evaluate the nature and extent of hearing loss and for eventual follow-up.
Histologic findings are nonspecific. The constricting bands consist of fibrous connective tissue resembling scars.
Management
Tailor medical care to individual defects or functional impairment of the limbs or hearing. Also see Medication.
A study presented in 2023 by Ling et al indicates that low-dose transforming growth factor β1 (TGF-β1) is a potential treatment. TGF-β1 expression is reduced in Vohwinkel syndrome with GJB2 mutations, and the in vitro study showed increased keratinocyte proliferation and cellular repair in Vohwinkel syndrome cells when TGF-β1 was introduced. [6]
Surgical release of the constriction bands is used to preserve the digits (eg, Z-plasty, other methods for relaxing scars). Surgical amputation is sometimes necessary due to pseudoainhum recurrence if autoamputation has not already taken place. [7]
No dietary interventions are indicated for treatment. Oral bioavailability of retinoids is enhanced with food intake.
Consultation with the following specialists may be needed:
-
Surgeon - If any deformities of the fingers or craniofacial features are present
-
Audiologist and speech therapist - If hearing loss is noted
-
Genetic counselor - Advisable to discuss risk of the offspring being affected by the syndrome
-
Other specialists - As indicated by clinical abnormalities
Regular follow-up by a dermatologist is recommended. Refer the patient to a surgeon if pseudoainhum develops. Further care and laboratory testing or radiography depend on the treatment selected, course, and complications. Audiologists and speech therapists may be referred on an individual basis.
Pathophysiology
Vohwinkel syndrome belongs to the group of palmoplantar keratodermas. It is considered to have an autosomal dominant inheritance, although sporadic cases [8, 9] and a case of probable autosomal recessive inheritance [10] have also been described.
Connexins 26, 30, 30.3, 31, and 43 are related to cutaneous diseases associated with multiple organ involvement. Mutations in connexin 26 are linked to Vohwinkel syndrome, keratitis-ichthyosis deafness and hystrixlike ichthyosis deafness syndromes, palmoplantar keratoderma with deafness, deafness with Clouston-like phenotype, and Bart-Pumphrey syndrome. [11]
Two mutations of the epidermal differentiation complex have been identified in Vohwinkel syndrome.
One is a missense mutation of the GJB2 gene coding connexin-26, a gap junction protein. [12, 13, 14, 15, 16] This mutation on chromosome 13 is associated with the classic (hearing loss–associated) Vohwinkel syndrome. Connexins are building blocks of gap junctions that are plasma membrane complexes facilitating and regulating the passage of small molecules between cells. Several other rare mutations have also been described.
Another mutation is an insertional mutation of the loricrin gene on the epidermal differentiation complex on 1q21. This protein plays a major function in the formation of the cornified cell envelope. Sequential deposition of altered loricrin during terminal differentiation of keratinocytes and other components causes an increase in envelope thickness and rigidity. A phenotype associated with ichthyosis and not deafness is observed.
An ichthyotic variant has been described with a 730insG mutation. [17]
Etiology
Causes of the two types are as follows:
-
Ichthyosis-associated type - Insertional mutation of the loricrin gene
-
Hearing loss–associated type - Missense mutation of the connexin-26 gene [13]
Epidemiology
The syndrome is rare, with fewer than 30 cases reported.
No racial or sex predominance is noted.
This syndrome usually manifests between infancy and early childhood.
Prognosis
The prognosis is good as long as medications are used. Patients with this syndrome may have a normal life span, persistent keratoderma, potential loss of digits, and hearing loss in the classic variant. Prenatal diagnosis by DNA analysis is possible if the gene defect is known. [18]
-
Starfish-shaped plaques.
-
Palmar hyperkeratosis.
-
Pseudoainhum.









