Background
Variegate porphyria (VP) is an inherited disorder of porphyrin-heme metabolism arising from mutations of the gene encoding the enzyme protoporphyrinogen oxidase. [1, 2, 3, 4] Manifestations of variegate porphyria in any given individual may include cutaneous photosensitivity (see image below), systemic symptoms arising from neurologic dysfunction, or both. [3, 5, 6, 7, 8]
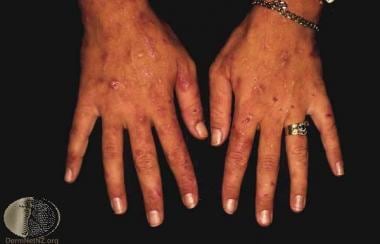 Variegate porphyria. Courtesy of DermNet New Zealand (http://www.dermnetnz.org/assets/Uploads/systemic/vp1.jpg0).
Variegate porphyria. Courtesy of DermNet New Zealand (http://www.dermnetnz.org/assets/Uploads/systemic/vp1.jpg0).
Pathophysiology
Inherited as an autosomal dominant trait, variegate porphyria is biochemically characterized by accumulations of the photosensitizing porphyrins, protoporphyrin and coproporphyrin. [5] In addition, abnormally high levels of the porphyrin precursors, porphobilinogen and aminolevulinic acid, are found during episodic attacks of systemic symptoms. [5, 9] Acute attack symptoms are caused by dysfunctions of central, autonomic, and peripheral nervous systems that appear to be effects of deranged heme synthesis on neurons. [10]
Etiology
Variegate porphyria arises from autosomal dominant inheritance of a gene mutation encoding a defective protoporphyrinogen oxidase enzyme. To date, there have been 184 different mutations in the protoporphyrinogen oxidase gene that results in variegate porphyria. [11] Individuals with one such mutation have total enzyme activity diminished to as much as 50% of normal. Most individuals with one mutation remain asymptomatic, but they are at risk if exposed to environmental factors (most often drugs) that can induce overt phenotypical expression. Systemic stress, such as liver disease or cholelithiasis, has been noted to precipitate cutaneous symptoms in patients with underlying variegate porphyria. [12]
Inheritance of two mutant protoporphyrin oxidase genes causes a more profound reduction in residual enzyme activity to 25% or less, leading to more severe disease manifestations. [13, 14] Eleven such cases have been identified at this time. These homozygous patients present during early childhood with developmental delay, nystagmus, photosensitivity, and skeletal hand deformities. [11]
The founder gene mutation common among South Africans encodes an enzyme with little or no residual activity that may be lethal in homozygotes. Four South African children manifesting variegate porphyria were compound heterozygotes with the founder mutation on one allele, but a different mutation apparently encoding an enzyme with enough residual activity to enable survival, on the other. [15]
Epidemiology
Frequency
United States
A registry has been established to develop information about the prevalence of porphyrias in the United States (American Porphyria Foundation;http://www.rarediseasesnetwork.org/cms/porphyrias/registry). Unknown numbers of cases may be unrecognized or misdiagnosed as another porphyria.
International
Prevalence is estimated at 1 case in 300 persons in South Africa, where a protoporphyrinogen oxidase gene "founder" mutation traceable to Dutch immigrants who married there in 1680 has been widely disseminated. [15, 16] It is the most common acute porphyria in Chile, [17] where another founder mutation was identified in four apparently unrelated families. [18] A haplotype study of Argentinean patients with variegate porphyria suggested a founder mutation within its population as well. [19] Among the combined populations of 11 European countries, 70 new cases of variegate porphyria were identified over a 3-year period; assuming mean disease duration of 40 years, prevalence in Europe was calculated at 3.2 cases per million persons. [20] In Israel, its prevalence was estimated to be 6.3 cases per million people. This relatively high prevalence compared with Europe suggests a possible protoporphyrinogen oxidase gene mutation passed down through Jewish ancestry. [21]
Race
No well-established racial predilection is known, although the disease is common among South Africans of Dutch ancestry, many of whom inherit the founder gene propagated in that population. Most multiple case reports involve European populations or North or South American populations that include individuals with European heritage.
Sex
Variegate porphyria occurs in both men and women, but its distribution between the sexes remains unclear. A study exploring its prevalence in Europe suggested a female predominance [20] ; however, an Israeli cohort study found an equal distribution between the sexes. [21] In 2015, a publication by Ramanujam and Anderson stated that variegate porphyria is more often symptomatic in women. [22]
Age
Variegate porphyria usually presents after puberty. Very rare childhood cases have been ascribed to the presence of 2 mutant protoporphyrinogen oxidase genes in the same individual. [13, 23]
Prognosis
Mild attacks of variegate porphyria may resolve within a few to several days with conservative management. Those that progress to vomiting or early signs of neuropathy usually respond to the administration of a heme analogue for 4 days. Profound attacks that are either unrecognized or inadequately treated early enough in the course may progress to long-term debility or death.
Cutaneous photosensitivity may cause difficulty performing manual labor and may limit many daily activities. Neurovisceral disorders vary from relatively mild symptoms that can remit spontaneously to profound crises that can be fatal or incapacitating for months to years. [3, 5, 6, 7, 24]
Acute attacks of variegate porphyria can be life-threatening. However, with early diagnosis of active disease, identification of asymptomatic protoporphyrinogen oxidase gene mutation carriers, and avoidance of drugs and other factors known to induce or worsen clinical expression, symptomatology among those at risk can be minimized. Good medical management of overt episodes can reduce morbidity and mortality such that the long-term outlook for patients is positive.
Patient Education
Patients need careful instruction about the nature of the disease and its genetic implications for their family members. Current lists of safe and unsafe drugs should be provided to patients and their physicians. Dietary instructions for maintaining adequate carbohydrate intake and for consuming a rapidly absorbed form of glucose at the first signs of an attack should be given. Effective sun avoidance practices should be advised for those patients with cutaneous photosensitivity.
-
Variegate porphyria. Courtesy of DermNet New Zealand (http://www.dermnetnz.org/assets/Uploads/systemic/vp1.jpg0).




