Background
A neurilemmoma, also known as schwannoma, neurolemmoma, and peripheral fibroblastoma, is a benign, encapsulated neoplasm derived from Schwann cells. Along with neurofibroma, schwannoma constitutes one of the two most common benign peripheral nerve sheath tumors. The peripheral nervous system can be defined as nervous tissue outside the brain and spinal cord. It extends from the glial-schwannian junction in the cranial nerves and spinal roots to the termination of the nerve fibers in their end-organ receptors and includes the posterior root ganglia and those of the autonomic nervous system. Neurilemmomas may affect any location in the course of the peripheral nervous system (ie, cranial and spinal nerve roots, cranial and peripheral nerves, end-organ receptors, small nerve twigs). They are common in paravertebral locations and the flexor regions of the extremities (especially near the elbow, wrist, and knee) and occasionally involve the skin. The presence of a noninvasive tumor next to a peripheral nerve suggests the diagnosis of neurilemmoma.
The major forms of neurilemmoma recognized are the conventional (common, solitary), cellular, plexiform, ancient forms, and melanotic schwannoma. [1, 2, 3] Specific variants such as plexiform and giant sacral neurilemmomas have been associated with an increased risk of local recurrence following incomplete excision.
Rare variants include microcystic/reticular, [4] benign and malignant epithelioid, [5, 6] pseudoglandular, [7, 8] neuroblastoma-like/rosetoid, [9] and a variant with a plexiform multinodular pattern. [10]
Other variants are associated with genetic syndromes such as Carney complex (ie, psammomatous melanotic schwannoma); neurofibromatosis (NF) type 2 (NF2, ie, cranial or spinal root neurilemmoma; multiple cutaneous plexiform schwannoma [11] ; neurilemmomatosis (schwannomatosis), which is a variant of NF2 characterized by multiple neurilemmomas and lack of involvement of the vestibular division of cranial nerve VIII; and segmental schwannomatosis, a distinct form of neurofibromatosis characterized by multiple schwannomas localized to one limb. [12] There have also been reports of nerve sheath tumors exhibiting histologic hybrid features of schwannoma and soft-tissue perineurioma. [13]
Of note, the benign nerve sheath tumors are classified as World Health Organization (WHO) grade I on the basis of their benign cytologic features, in contrast to the malignant counterparts, which are WHO grade III or IV.
Pathophysiology
An understanding of the relationship of the Schwann cell to other neuronal elements in the peripheral nervous system is helpful in conceptualizing the pathophysiology of a neurilemmoma.
The peripheral nervous system differs from the central nervous system in the nature of its supporting cell; the Schwann cell supports the former and the neuroglial cells support the latter. It is generally accepted that in embryogenesis, the Schwann cells are derived from the neural crest and are of neuroectodermal origin. As the peripheral nerves form, the Schwann cells migrate peripherally from the spinal ganglia, parallel to the axons, and encase them with their cytoplasm. In myelinated fibers, only one axon segment is encased by one Schwann cell.
The myelin sheath is created by a synthesis and wrapping of Schwann cell plasma membrane around the axon. Schwann cells sheath the axons from the point at which the latter penetrate the pia mater to their terminations. Upon penetrating the pia, the neuroglia is lost; the individual nerve fibers pass through a sieve-like structure composed of reticulin (young collagen fibers), and, thereafter, each is contained within its ensheathing tube of reticulin and Schwann cell elements.
At this site, perineurial cells (perineural epithelium) also make their appearance. In nonmyelinated nerves, several axon segments are ensheathed by a common Schwann cell. In a fully developed nerve, a layer of connective tissue, or epineurium, surrounds the entire nerve trunk. Several nerve fascicles lie within the confines of the epineurium, and each is surrounded by a well-defined perineurium. The smallest connective tissue unit of the nerve is the endoneurium, which is a network of fibroblasts, blood vessels, and collagen encircling individual nerve fibers).
Neurilemmomas arise within a nerve consisting of a single fascicle. The tumors are composed entirely of the supporting Schwann cells and peripherally displaced nerve fibers, resulting in a globoid eccentric tumor mass. In the early intrafascicular growth phase, small neurilemmomas displace nerve fibers without forming a capsule. The larger tumors increase the size of the parent nerve and become separated from surrounding fascicles by a capsule derived from perineurium and epineurium, as demonstrated in the image.
A schematic illustration of the essential microscopic features of a neurilemoma (schwannoma). A solid lesion arises within a nerve composed of a single fascicle (top). The tumor is composed of Schwann cell proliferation within the epineurium and peripherally displaced nerve fibers, resulting in nodular eccentric growth (middle). No capsule is formed in the early growth phase. The larger tumor (bottom) slightly increases the size of the parent nerve and eventually becomes separated from surrounding fascicles by a capsule formed from the perineurium and epineurium. Occasional axons are present.
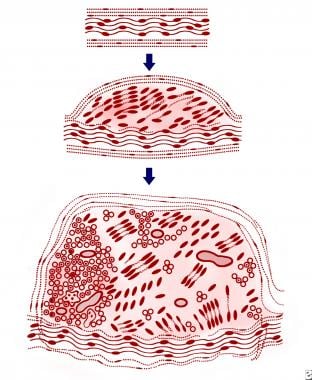 A schematic illustration of the essential microscopic features of a neurilemoma (schwannoma). A solid lesion arises within a nerve composed of a single fascicle (top). The tumor is composed of Schwann cell proliferation within the epineurium and peripherally displaced nerve fibers, resulting in nodular eccentric growth (middle). No capsule is formed in the early growth phase. The larger tumor (bottom) slightly increases the size of the parent nerve and eventually becomes separated from surrounding fascicles by a capsule formed from the perineurium and epineurium. Occasional axons are present.
A schematic illustration of the essential microscopic features of a neurilemoma (schwannoma). A solid lesion arises within a nerve composed of a single fascicle (top). The tumor is composed of Schwann cell proliferation within the epineurium and peripherally displaced nerve fibers, resulting in nodular eccentric growth (middle). No capsule is formed in the early growth phase. The larger tumor (bottom) slightly increases the size of the parent nerve and eventually becomes separated from surrounding fascicles by a capsule formed from the perineurium and epineurium. Occasional axons are present.
Most neurilemmomas are of the conventional (common) type, arise as solitary tumors smaller than 10 cm, and are not associated with a genetic syndrome. They display the classic gross and microscopic features described in the Histologic Findings. The cellular variant is rare in the skin, developing more commonly as a tumor of the mediastinum, retroperitoneum, and deep soft tissue.
In contrast to plexiform neurofibroma, plexiform neurilemmoma is not pathognomonic of neurofibromatosis and malignant transformation is exceedingly rare.
Etiology
Solitary neurilemmomas are sporadic in 90% of the time or they may occur as part of a familial syndrome. [14] Most tumors, whether solitary or part of a syndrome, have shown genetic aberrations (ie, ring chromosome 22). [15]
The NF2 gene has been localized to band 22q12. Alteration or loss of the NF2 gene product (also designated as Merlin), a presumed tumor suppressor gene, is central to the pathogenesis of these tumors. [14, 16] Partial or complete monosomy of the chromosome occurs (ie, loss or mutation of both NF2 alleles and mutation of the NF2 gene protein).
The negative staining of neurilemmoma cells by immunohistochemical stain for the NF2 protein suggests that loss of NF2 protein function is a prerequisite for neurilemmoma formation.
A neurofibroma/schwannoma hybrid was recognized by the WHO Classification of Tumours of the Central Nervous System, due to an ERBB2 mutation. [17]
Melanotic schwannoma is associated with a mutation in the PRKAR1A gene and the Carney complex. [18]
More than 150 cases of radiation-induced intracranial and peripheral neurilemmomas have been reported. [19] The mean latency period is approximately 20 years, and most of these are solitary tumors.
Epidemiology
Frequency
The exact prevalence of neurilemmomas (benign schwannomas) of all anatomic sites is unknown. It is estimated that about 70% of NF2 patients have skin tumors, the majority of which are schwannomas. [20] Meanwhile, the plexiform variant of this Schwann cell tumor was found to have a 5% association with NF2 and schwannomatosis. [21] Numerous subsequent reports have confirmed that this association is more than coincidental.
Race
No racial predilection is noted for this neoplasm.
Sex
The tumor affects the sexes in roughly equal numbers. [22]
Age
Neurilemmomas occur in persons of any age but are most common in patients aged 20-50 years. The cellular form of neurilemmoma has a peak incidence in the fourth decade of life, and approximately 5% of neurilemmomas occur in childhood and adolescence. Three congenital cases have been reported. In the nearly 50 reported cases of plexiform neurilemmoma, the age ranged from 50 days to 80 years, with a mean of 34 years. In melanotic neurilemmoma, the age range was reported at 10-84 years, with a mean age of 37 years. Overall, approximately 75% of neurilemmomas occur in the first 4 decades of life.
Prognosis
Neurilemmomas behave in a benign fashion. Incompletely excised examples are capable of slow recurrence. Higher recurrent rates are noted with the plexiform and cellular varieties. [23] Locally aggressive behavior is observed in tumors with increased cellularity, higher mitotic rates (mean, 4 per 10 high-power fields), and underlying bone extension (observed in occasional cases of orbital neurilemmoma).
Melanocytic schwannomas of the cervical, thoracic, and lumbar spine reported by Peltier et al [24] demonstrated a guarded prognosis. Two of the three cases studied showed unfavorable outcomes, with local recurrence and leptomeningeal metastasis, especially in young patients.
Large tumors (eg, giant sacral schwannomas), contrary to previous literature, have been found to have a low rate of local recurrence and rare malignant transformation. [25] A clinicopathologic study has found that patients with asymptomatic neurilemmomas occurring in association with NF2 not only have more severe neurologic deficits but also have little postoperative improvement and a higher rate of tumor recurrence. Malignant schwannomas (neurofibrosarcoma) arise de novo or in neurofibromas in the setting of neurofibromatosis. They are not regarded to be malignant counterparts of neurilemmomas. Malignant change in neurilemmomas is exceedingly rare.
-
A schematic illustration of the essential microscopic features of a neurilemoma (schwannoma). A solid lesion arises within a nerve composed of a single fascicle (top). The tumor is composed of Schwann cell proliferation within the epineurium and peripherally displaced nerve fibers, resulting in nodular eccentric growth (middle). No capsule is formed in the early growth phase. The larger tumor (bottom) slightly increases the size of the parent nerve and eventually becomes separated from surrounding fascicles by a capsule formed from the perineurium and epineurium. Occasional axons are present.
-
A small, clinically freely movable neurilemoma found in the subcutaneous tissue. Note the pale-yellow, somewhat-translucent cut surface. The tumor also exhibits a slight nodular growth pattern on the cut surface. Courtesy of the Atlas of Tumor Pathology Armed Forces Institute of Pathology Fascicles, Tumors of the Peripheral Nervous System. Used with permission.
-
A larger neurilemoma (5 cm in diameter) arising from a peripheral nerve showing irregularly lobulated and secondary degenerative changes, ie, partly cystic with calcification (the so-called ancient change). Hemorrhage and opaque creamy-yellow areas of tumor are also seen on this cut surface.
-
Cut surface of an intradermal plexiform (nodular) variety of neurilemoma. The plexiform variants of neurilemoma are rare. The area of nodularity is clearly discernible. Courtesy of the Atlas of Tumor Pathology Armed Forces Institute of Pathology Fascicles, Tumors of the Peripheral Nervous System. Used with permission
-
A low-power photomicrograph of a dermal plexiform neurilemoma showing nodular aggregates of tumor cells and surrounding loose, myxomatous fibrous stroma. Hematoxylin and eosin stain at 50X magnification.
-
Photomicrograph of a neurilemoma from an area with a typical Antoni type A pattern. The palisaded benign Schwann cells show nuclear crowding, with cell processes radiating toward the centers of aggregated tumor cells. Inconspicuous loose fibrous stroma is present at the periphery. Hematoxylin and eosin stain at 150X magnification.
-
A photomicrograph showing a characteristic Verocay body of a neurilemoma, consisting of tight, discrete aggregates of spindle-shaped, palisaded nuclei with a central fibrillary area, representing collections of cytoplasmic processes of tumorous Schwann cells. Courtesy of the Atlas of Tumor Pathology Armed Forces Institute of Pathology Fascicles, Tumors of the Peripheral Nervous System. Used with permission.
-
Transmission electron micrograph of Antoni type A tumor tissue consisting of prominent arrays of Schwann cell processes with basement membrane substance coated on their surfaces. Note the centrally located nucleus with vesicular nuclear chromatin. Uranium acetate and lead citrate stain at 15,000X magnification.
-
A transmission electron micrograph of a Luse body, ie, typical collagen fibrils and adjacent basement substance. Note the long-spaced, 130-nm periodicity. Uranium acetate and lead citrate stain at 52,500X magnification. Courtesy of the Atlas of Tumor Pathology Armed Forces Institute of Pathology Fascicles, Tumors of the Peripheral Nervous System. Used with permission.
-
A photomicrograph of a dermal neurilemoma with anti–S-100 protein immunostaining. The tumorous Schwann cells exhibit uniformly positive staining. Immunoperoxidase stain at 150X magnification.
-
Solitary cutaneous plexiform neurilemoma shown on photomicrograph.
-
Management algorithm for schwannoma nerve graft.






