Background
Acquired angioedema (AAE) due to acquired C1-inhibitor (C1-INH) deficiency (C1-INH-AAE) is a rare disorder caused by acquired consumption of C1-INH. It is clinically characterized by recurrent episodes of nonpitting asymmetric edema of the face, lips, tongue, limbs, and genitals; severe abdominal pain due to edema of the gastrointestinal mucosa; and life-threatening edema of the upper respiratory tract. It is not associated with urticaria and is not an immunoglobulin E (IgE)‒mediated process.
Acquired angioedema was first described by Caldwell et al in 1972. The three key elements that initially characterized acquired angioedema were acquired deficiency of C1-INH, hyperactivation of the classic pathway of human complement, and recurrent angioedema symptoms. [1]
Two distinct syndromes are described below. Both types of acquired angioedema (AAE) without urticaria are characterized by painless, nonpruritic, nonpitting swelling of the skin. They are classified into 2 forms: acquired angioedema type I (AAE-I) and acquired angioedema type II (AAE-II).
Acquired angioedema type I is associated with other diseases, most commonly B-cell lymphoproliferative disorders. Acquired angioedema type II is an autoimmune process defined by the presence of an autoantibody directed against the C1 inhibitor molecule (C1-INH).
See the image below.
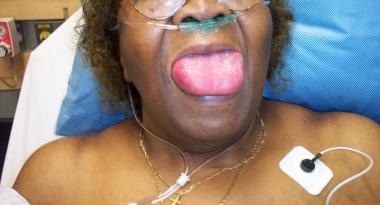 Angioedema secondary to ACE inhibitors.
Angioedema secondary to ACE inhibitors.
Go to Angioedema, Pediatric Angioedema, Acute Angioedema, and Hereditary Angioedema for complete information on this topic.
Pathophysiology
C1-INH is a multifunctional serine protease inhibitor that is normally present in high concentrations in plasma. It is primarily synthesized by hepatocytes. Its synthesis is up-regulated by interferon-gamma, interleukin 6, and interleukin 1. Androgens may also have a role in stimulating C1-INH synthesis. The major functions of C1-INH include inhibition of activated C1r and C1s, inhibition of activated Hageman factor (XIIa), and inhibition of activated kallikrein, the contact system protease that cleaves kininogen and releases bradykinin. [2]
Bradykinin is an important mediator involved in tissue permeability and vascular dilatation. Its biological effect is exerted through activation of bradykinin B2 receptors, which are expressed in the membranes of endothelial and smooth muscles cells. Elevated blood bradykinin levels are found during clinical flares in patients with angioedema. Other kinins may also be pathogenic. [2]
The specific trigger responsible for inducing the release of these vasoactive peptides is unclear. Activation of factor XII (Hageman factor) may be secondary to phospholipid release from damaged or apoptotic cells and may be important in the generation of bradykinin from endothelial activation. This hypothesis encompasses the role of illness or tissue injury in the generation of bradykinin.
Supporting the importance of bradykinin in acquired angioedema, vascular permeability has been shown to increase in mice deficient in C1-INH, but not in mice with a deficiency in both C1-INH and the bradykinin B2 receptor. [3] The precise pathophysiology of acquired angioedema type I remains to be defined. C1-INH levels diminish as a result of its increased catabolism and excessive activation of the classic complement pathway.
Although the current classification of acquired angioedema is being readdressed, in acquired angioedema type I, the associated disorders (usually lymphoproliferative malignancies) produce complement-activating factors, idiotype/anti-idiotype antibodies, or other immune complexes that destroy C1-INH function. Neoplastic lymphatic tissue has been found to play an active role in the consumption of C1-INH and the complement components of the classic pathway.
The most commonly associated malignancy, B-cell lymphoma, has shown that anti-idiotypic antibody attached to immunoglobulin on the surface of B-cells causes C1-INH deficiency. Increased consumption of C1q followed by C2 and C4 results in subsequent release of vasoactive peptides that act on postcapillary venules.
In acquired angioedema type II, a normal 105-kd C1-INH molecule is synthesized in adequate amounts but, because of an unknown event, a subpopulation of B cells secretes autoantibodies to the C1-INH molecule. This autoantibody, which may be of any of the major immunoglobulin classes, binds to the reactive center of C1-INH. After binding to C1-INH and altering its structure, its regulatory capacity is diminished or abrogated.
In all reported cases of C1-INH deficiency caused by an autoantibody, C1-INH circulates in the blood in a form that has been cleaved by target proteases from its native molecule to a 95-kd fragment. Because of the higher affinity of the autoantibody for native C1-INH, the 95-kd antibody/C1-INH complex dissociates, and the freed antibody can bind to another native C1-INH molecule, further depleting C1-INH.
The distinction between acquired angioedema type I and acquired angioedema type II may be difficult to make at times, and it is imperative to stress that overlap does occur. For instance, cases of monoclonal gammopathy of undetermined significance (MGUS) have shown the monoclonal immunoglobulin itself to be the C1-INH antibody. Patients with acquired angioedema type I may initially present with autoantibodies to C1-INH, or the autoantibodies may develop as the disease progresses. While measuring antibodies against C1-INH is useful, groups have also developed assays that can measure the antibody against C1-INH in complex with C1-INH. In one study, anti–C1-INH antibodies were detected in 9 of 20 patients, while the complex between anti–C1-INH and C1-INH was found in 18 of 20, suggesting that there could be greater numbers of patients with anti–C1-INH antibodies than is seen when measuring antibody alone. [4]
Etiology
Acquired angioedema type I is most frequently associated with B-cell lymphoproliferative disease. To date, only three reports of a T-cell lymphoma associated with acquired angioedema type I have been documented. [5]
Other associated disorders have included the following:
-
MGUS
-
Rectal carcinoma
-
Essential cryoglobulinemia
-
Erythrocyte sensitization
-
Cold urticaria
-
Lupus anticoagulant
-
Infection with Helicobacter pylori or Echinococcus granulosis
Acquired angioedema type II is not associated with any specific disorder but rather is characterized by the presence of autoantibody directed against C1-INH. Most of these antibodies work by binding the epitopes around the reactive center of INH. [6] However, the occasional existence of features of both acquired angioedema type I and acquired angioedema type-II has been noted, most notably with a MGUS.
A 2016 study reported that 33% of patients presenting with acquired angioedema had or would develop non-Hodgkin lymphoma, in particular marginal zone lymphoma. Of the studied patients, 62.5% were diagnosed with non-Hodgkin lymphoma at the onset of angioedema or up to 7 years later. Patients presenting with acquired angioedema and C1-INH deficiency should be monitored regularly for the development of malignancy. [7]
In addition to associations with MGUS, C1-INH–deficient AAE has also been reported in monoclonal gammopathy of renal significance (MGRS). [8] One case of acquired angioedema with C1-INH deficiency state was identified in association with liver transplantation. The status of the liver donor was unknown, but it is speculated that the donor may have been C1-INH deficient. Another case of acquired angioedema was reported with acute upper airway angioedema in association with the local anesthetic articaine.
Epidemiology
Acquired angioedema is rare. Only approximately 150 cases have been reported in the medical literature worldwide.
Racial and sexual differences in incidence
Persons of any race can be affected by acquired angioedema. Men and women are equally affected.
Age-related differences in incidence
The onset of acquired angioedema is most common after the fourth decade of life, whereas the usual onset of hereditary acquired angioedema (HAE) is in the second decade.
Prognosis
The prognosis for patients with acquired angioedema is variable, and, in most cases, it depends on control of the underlying disorder. However, even with appropriate treatment of the underlying disease, patients may be free of symptoms only temporarily. Additionally, some patients who have had their underlying disease treated will be free of symptoms related to angioedema but will continue to have biochemical abnormalities. [9]
Compared with the general population, patients with acquired angioedema have a higher incidence of B-cell malignancies. Patients with acquired angioedema and a concurrent diagnosis of monoclonal gammopathy of undetermined significance (MGUS) do not have an increased risk for progression to malignancy compared with patients with a sole diagnosis of MGUS.
Although mortality may occur because of laryngeal edema, it is more likely due to the complications of the associated disorder.
Patient Education
For patient education information, see the Allergy Center and Hives and Angioedema.
-
Angioedema secondary to ACE inhibitors.