Background
Juvenile xanthogranulomas (JXGs) are benign; usually asymptomatic; self-healing; red, yellow, or brown papules and nodules composed of histiocytic cells that predominantly occur in infancy and childhood. Papules or nodules occur in the skin, eyes, and viscera. JXG is the most common form of non–Langerhans cell histiocytosis. [1, 2]
See the image below.
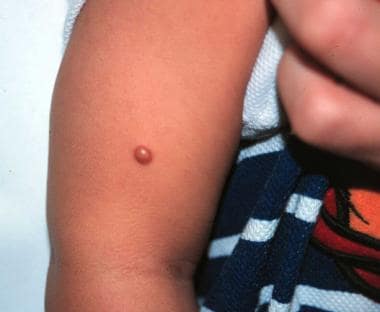 Smooth, domed, yellow-brown, 5-mm papule on right arm of a 6-month-old boy.
Smooth, domed, yellow-brown, 5-mm papule on right arm of a 6-month-old boy.
Adamson first reported JXG in the English literature in 1905. He presented a child who developed numerous yellow-white papules on the body in the first 2 weeks of life. He named the entity congenital xanthoma multiplex.
In 1912, McDonagh presented the first case review and renamed the condition nevoxanthoendothelioma (although the condition is not associated with nevi or endothelial cells). In 1954, Helwig and Hackney again retermed it juvenile xanthogranuloma, reflecting its histopathologic appearance. Laurb and Lain first reported JXG with visceral involvement in 1937. Blank et al first described ocular involvement in 1949.
Pathophysiology
The etiology of juvenile xanthogranuloma (JXG) is not fully known. The papules and nodules of JXG represent collections of differentiated non–Langerhans cell histiocytes. The consensus is that the cells of origin are dermal dendrocytes. As postulated, JXG may be a granulomatous reaction of histiocytes to an unidentified stimulus, possibly of either physical or infectious etiology. Evidence from Kraus et al, [3] however, suggests a possible CD4+ plasmacytoid monocyte origin. Inhibition of cellular apoptosis appears to play a minor role in the growth of xanthogranulomas. [4]
The appearance of giant cells and foamy lipid-laden histiocytes generally occurs late and apparently is a secondary event, possibly in response to cytokine production by histiocytes. Serum lipid levels are normal and remain normal.
Etiology
Coexistence of café au lait macules and juvenile xanthogranuloma (JXG) has been associated with epilepsy.
Niemann-Pick disease has been associated with JXG.
Urticaria pigmentosa has been associated with JXG.
Neurofibromatosis type 1 (NF1) has been associated with JXG. [5, 6, 7, 8, 9] A recent study found a prevalence of 37.5% of JXGs in young children with NF1. [10] Another recent study also found a high frequency (30%) of JXGs in children with NF1 who were younger than 2 years. [11] These JXG lesions were often multiple. It has been proposed that JXG may be a helpful diagnostic criterion for NF1.
Juvenile chronic myelogenous leukemia, now primarily referred to as juvenile myelomonocytic leukemia (JMML), has been observed in association with multiple JXGs, [7, 8, 12] and the prevalence is especially high in patients with coexistent neurofibromatosis. Statistics regarding this triple association are controversial; estimates indicate that patients with NF1 and JXG have a 20- to 32-times increased risk of developing JMML than patients with NF1 alone. Patients have also been diagnosed with JMML and JXG, but without NF1.
Epidemiology
Race
Juvenile xanthogranuloma (JXG) occurs in whites approximately 10 times more frequently than in African Americans.
Sex
In childhood, juvenile xanthogranuloma (JXG) occurs predominately in males (1.5:1). Equal incidence occurs in adult males and females. Multiple cutaneous lesions occur predominantly in males (12:1).
Age
Approximately 35% of cases of juvenile xanthogranuloma (JXG) occur at birth, with as many as 71% of cases occurring in the first year. The mean age at presentation is 22 months. Most JXGs resolve by age 5 years. Despite the term juvenile in the disease name, 10% of cases manifest in adulthood.
Prognosis
In the absence of therapeutic intervention, juvenile xanthogranulomas (JXGs) flatten with time. Both cutaneous and extracutaneous lesions involute spontaneously within 3-6 years.
Hyperpigmentation, mild atrophy, or anetoderma may persist.
Lesions can recur after resection. The relapse rate is approximately 7%.
In the absence of neurofibromatosis, no systemic health implications are involved, with a few rare exceptions.
Vigilantly screen patients with neurofibromatosis and JXG for leukemia.
Ocular, neurologic, and hepatic disease are rare but may have serious long-term consequences.
Patient Education
Reassure patients and their families. Instruct patients concerning associations related to clinical situations (neurofibromatosis, ocular findings in diffuse JXG, JMML), and direct patient education toward these conditions.
-
Smooth, domed, yellow-brown, 5-mm papule on right arm of a 6-month-old boy.





