Practice Essentials
Idiopathic pulmonary fibrosis (IPF) is a form of idiopathic interstitial pneumonia that is associated with a median survival of 2-5 years from the time of diagnosis. IPF affects mainly the elderly and is indicative of a link between a fibrotic process and aging. IPF is the most common and progressive fibrosing form of interstitial lung disease (ILD) and eventually leads to respiratory failure and decreased lung capacity, with a poor patient prognosis. Treatment can delay the progression of IPF, so early diagnosis is fundamental. [1, 2] Increasing awareness of the clinical manifestations of IPF, more widespread use of computed tomography (CT) scans, and other potential factors have contributed to the rising prevalence of IPF, especially in people older than 65 years. [3]
Usual interstitial pneumonia (UIP) is the hallmark the radiologic pattern of IPF and is divided into 4 categories: typical UIP, probable UIP, indeterminate for UIP, or suggestive of an alternative diagnosis. In cases of typical UIP, there is a subpleural and basal distribution of fibrosis characterized by honeycombing, with or without peripheral traction bronchiectasis or bronchiolectasis, and the absence of findings suggesting a diagnosis other than IPF. A probable UIP pattern lacks honeycombing but otherwise shows the features of a typical UIP pattern. [4, 5]
Classic features of IPF include progressive dyspnea and nonproductive cough. Pulmonary function tests usually reveal restrictive impairment and diminished carbon monoxide–diffusing capacity. Diagnosis can be made without biopsy with compatible imaging tests, appropriate clinical history, and exclusion of other conditions. The classic pattern of imaging on CT shows peripheral distribution of bilateral fibrosis, more pronounced at the bases. If diagnosis is uncertain, IPF can be diagnosed by lung biopsy. [6]
It is theorized that molecular mechanisms causing IPF reflect an abnormal reparative response to repeated alveolar epithelial damage in an aging, genetically sensitive individual. One goal of current research is to develop early biomarkers for IPF that may include circulating variables, demographic information, and imaging data. [7]
(Radiologic characteristics of pulmonary fibrosis are shown in the image below.)
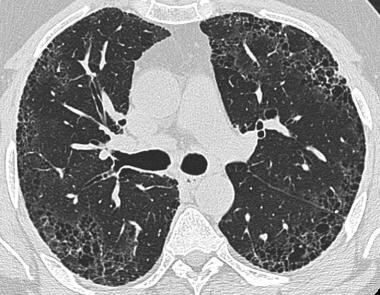 HRCT of advanced stage of pulmonary fibrosis demonstrating reticular opacities with honeycombing, with predominant subpleural distribution.
HRCT of advanced stage of pulmonary fibrosis demonstrating reticular opacities with honeycombing, with predominant subpleural distribution.
Diagnosis is confirmed with a lung biopsy, but histology shows striking variation from one region to the next (ie, the disease is characterized by histologic temporal and spatial heterogeneity). It is not unusual to find areas of normal lung next to areas with severe thickening of alveolar walls. Therefore, findings on bronchoscopic or percutaneous lung biopsy are difficult to interpret. Open lung biopsy and video-assisted thoracoscopic lung biopsy are preferred methods.
IPF usually affects people 50-70 years of age. Most series report a male preponderance, with a male-to-female ratio of 2:1. Clinical features consist of progressive exertional dyspnea; the presence of interstitial infiltrates, as evidenced on chest radiographs; and physiologic evidence of restriction and impaired gas exchange on pulmonary function testing.
Patients generally are treated with corticosteroids, other immunosuppressants, or both.
Imaging modalities
Idiopathic pulmonary fibrosis is diagnosed on the basis of patient history, clinical findings, pulmonary physiology, and imaging results. The diagnosis is one of exclusion. Nonidiopathic causes must be excluded first because of important therapeutic implications. After nonidiopathic causes are excluded, further investigation of patients with IPF typically reveals radiographic abnormalities and restrictive lung physiology with decreased diffusion capacity. [8, 9, 10]
Plain chest radiography is usually the first investigation performed for patients with suspected interstitial lung disease. However, findings on conventional radiography are highly nonspecific.
High-resolution CT (HRCT) is currently the method of choice for diagnosis of IPF. However, HRCT in a typical usual interstitial pneumonia (UIP) provides a definitive answer in just over 50% of patients, thereby necessitating invasive tissue diagnosis. [11] HRCT defines underlying lung parenchymal abnormalities better than plain radiography. [12, 13] The classic pattern of imaging on CT shows a peripheral distribution of bilateral fibrosis, more pronounced at the bases. [6] Studies have shown that HRCT may obviate surgical lung biopsy in some patients.
Raghu et al compared the diagnostic accuracy of clinical evaluation in combination with HRCT with the accuracy of histology of surgical lung biopsy samples. [14] Clinical assessment in conjunction with careful review of HRCT scans was 60% sensitive and 97% specific for IPF. However, although HRCT may obviate the need for tissue diagnosis in 60% of patients, surgical lung biopsy is still needed in 40%.
For diagnoses other than IPF, a combination of clinical assessment and HRCT is neither sensitive nor specific enough to be relied on without surgical biopsy. Open lung biopsy remains the criterion standard. For immunocompetent patients, the benefit is relatively low because corticosteroid therapy is frequently administered after biopsy. For immunocompromised patients, approaches to therapy change substantially after tissue confirmation, but mortality is high. Therefore, open biopsy should be performed only in patients for whom the diagnosis is likely to change therapy and in patients who have a reasonable prognosis.
Radionuclide scanning with gallium-67 may depict interstitial fibrosis and may show changes early. This feature may be of therapeutic benefit, but changes are nonspecific and do not remove the need for lung biopsy.
Lung ultrasound (LUS) is a noninvasive, radiation-free, well-tolerated, sensitive technique for detection of early changes of fibrotic lung disease. LUS can be used to monitor disease progression in the natural course or after treatment initiation. LUS is portable and can be performed at the patient’s bedside. [11]
Dynamic contrast-enhanced MRI can detect changes in the microvasculature and extravascular extracellular space in IPF, thus providing in vivo regional functional information. [15]
Guidelines
Fleischner Society
The Fleischner Society White Paper details the expanded role of CT to allow diagnosis of IPF without surgical lung biopsy when CT shows a probable pattern of usual interstitial pneumonia (UIP). They note that additional investigations, including surgical lung biopsy, should be considered in patients with either clinical or CT findings that are indeterminate for IPF. They specifically noted the following [16] :
-
The typical UIP pattern is characterized by reticular opacities with obligatory honeycombing, usually associated with traction bronchiectasis.
-
Ground-glass opacity, if present, is usually admixed with reticular abnormality and honeycombing. Such abnormalities are characteristically basal and peripheral, though often patchy.
-
Some degree of upper lung involvement (including honeycombing) is usual, and sometimes the craniocaudal distribution may be relatively uniform in patients with otherwise typical UIP.
-
Optimal-quality CT requires the use of thin section (< 2mm) and high spatial resolution reconstruction.
-
Images should be obtained at full inspiration to total lung capacity.
-
Volumetric CT acquisition is preferred to noncontiguous imaging.
-
On CT, honeycombing is defined as clustered, thick-walled, cystic spaces of similar diameters, generally measuring 3-5 mm, but occasionally up to 25 mm.
Radiology Working Group
The Radiology Working Group of the Pulmonary Fibrosis Foundation stated that in cases in which the CT pattern suggests typical or probabey UIP, if the clinical scenario is consistent with IPF, biopsy is unnecessary. However, biopsy is necessary if the clinical scenario is not typical of IPF (eg, patient younger than 60 yr, relevant inhalational exposures). [17, 18, 16, 4]
ATS/ERS/JRS/ALAT
The American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society in a collaborative effort made the following recommendations for IPF [5] :
-
UIP is the hallmark radiologic pattern of IPF. 4 diagnostic categories of UIP were created based on HRCT features: a UIP pattern, a probable UIP pattern, an indeterminate for UIP pattern, and alternative diagnosis.
-
HRCT features frequently seen in UIP include honeycombing, traction bronchiectasis, and traction bronchiolectasis, which may be seen along with ground-glass opacification and fine reticulation. Honeycombing is a distinguishing feature of UIP and must be present for a definite HRCT diagnosis of UIP to be made.
-
Subpleural, basal-predominant reticular abnormalities with peripheral traction bronchiectasis or bronchiolectasis should be regarded as probable UIP. As with a UIP pattern, ground-glass opacification may be present in probable UIP, but it is not a dominant feature.
-
Indeterminate for UIP pattern should be assigned when HRCT demonstrates features of fibrosis but does not meet UIP or probable UIP criteria and does not explicitly suggest an alternative diagnosis.
-
The diagnostic approach to IPF is highly reliant on images of the lungs generated from volumetric scanning of the chest. This mode has essentially replaced sequential CT scanning because it improves detection of all abnormalities, even if subtle or focal.
-
Technical requirements of HRCT include the following: thinnest collimation, shortest rotation time, and highest pitch to ensure creation of motion-free images. The first acquisition is obtained in the supine position at sustained end-inspiration (volumetric acquisition). The second acquisition is obtained in the supine position over the entire thorax at sustained end-expiration, after a prolonged expiration (volumetric or sequential acquisition). The third acquisition is aimed at clearing position-induced changes in the dependent lung of the first acquisition.
-
Chest CT angiography should be obtained to detect pulmonary embolus
Radiography
The radiographic pattern of IPF differs with disease stage. Early in the disease, the most common radiographic changes include an interstitial shadowing of small (1-2 mm), irregular opacities, which are seen in about 75% of patients. Less common are small, round opacities, which are seen in 20% of patients. This finding is generally known as reticulonodular opacities. Septal lines occasionally are observed, and the distribution is predominantly basal. (See the image below.)
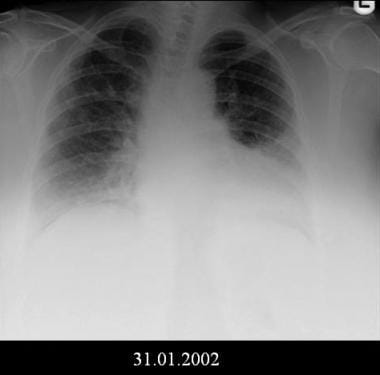 Bilateral lower lobe opacities and possible mild decrease in lung volumes. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
Bilateral lower lobe opacities and possible mild decrease in lung volumes. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
Peripheral accentuation is a common feature, but this is more easily appreciated on CT than on plain chest radiography.
The pattern is usually symmetrical. Another common pattern is hazy, ground-glass opacification, which may be diffuse or patchy. Volume loss and a raised diaphragm are seen in up to 60% of patients. These may be accompanied by basal discoid atelectasis.
Pleural disease is not typical of IPF. Its presence should raise the possibility of other conditions, such as asbestosis, rheumatoid pulmonary disease, or systemic lupus. Pneumothorax, pneumomediastinum, or both have been reported in a few patients; these conditions have been associated with bullae in the lung parenchyma.
With progression of alveolitis to fibrosis, initial fine lines become coarse and small (2 mm) cysts appear. These cysts coalesce and increase to 5-7 mm in diameter; they appear as ring opacities within the honeycomb lung. As fibrosis worsens, honeycombing becomes coarser with larger honeycomb cysts, and further volume loss is evident. At advanced stages, radiographic evidence of pulmonary arterial hypertension is seen.
Degree of confidence
Radiographic findings are not correlated with stage of disease, histology, respiratory symptoms, respiratory function tests, or prognosis.
In most patients with IPF, the chest radiograph is abnormal at presentation; often, previous radiographs show reticular shadowing, even before symptoms develop. [19] Chest radiography frequently is the first investigation employed for patients with IPF; physiologic testing and HRCT scanning follow.
For symptomatic patients for whom diffusion capacity is abnormal, results of chest radiography may be normal. For other patients, radiographic appearances are abnormal before clinical symptoms appear. Results of HRCT scanning are abnormal for most patients with IPF.
Computed Tomography
For patients with interstitial pulmonary fibrosis (IPF), high-resolution computed tomography (HRCT) findings may be used to predict outcomes and to guide treatment, because findings are well correlated with the histologic pattern of IPF (see the images below). Accuracy of the diagnosis of IPF is significantly increased with HRCT, as compared to chest radiography. When a trained observer performs HRCT, the accuracy of the diagnosis is reported to be about 90%. [20] One third of all cases of IPF are missed on HRCT; a confident diagnosis of IPF is made in about two thirds of cases. [20]
On HRCT, end-stage lung disease is characterized by honeycombing without ground-glass attenuation in typical distribution; with such findings on HRCT, the diagnosis may be made with confidence. This spares patients the risk of invasive diagnostic processes such as lung biopsy. In the active stage, scans reveal ground-glass attenuations. The active stage of disease, which is characterized by active alveolitis, is potentially reversible and potentially amenable to treatment, unlike end-stage disease, which is irreversible. [21, 22, 23, 24]
HRCT of advanced stage of pulmonary fibrosis demonstrating reticular opacities with honeycombing, with predominant subpleural distribution.
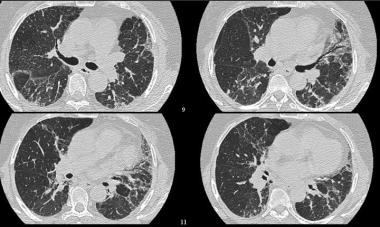 High-resolution CT (HRCT) shows increased pulmonary attenuation with distortion of the pulmonary architecture. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
High-resolution CT (HRCT) shows increased pulmonary attenuation with distortion of the pulmonary architecture. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
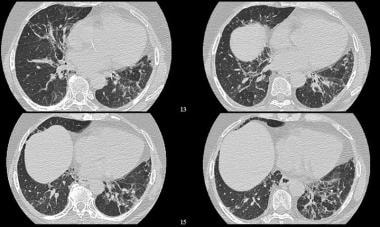 High-resolution CT (HRCT) shows distortion of the pulmonary architecture with thickening of pulmonary interstitium and some areas of ground-glass attenuation. No obvious honeycombing is present. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
High-resolution CT (HRCT) shows distortion of the pulmonary architecture with thickening of pulmonary interstitium and some areas of ground-glass attenuation. No obvious honeycombing is present. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
Interstitial B-lines, considered the hallmark of interstitial lung disease, are seen as vertical, hyperechoic artifacts. A thick, irregular, fragmented pleura line is associated with subpleural fibrotic scars. The total number of B-lines is correlated with extension and severity of pulmonary fibrosis on HRCT. The average distance between 2 adjacent B-lines is an indicator of a particular pattern on HRCT. This is used to appreciate a pure reticular fibrotic pattern in IPF, as compared to the predominantly ground-glass pattern seen in fibrotic nonspecific interstitial disease. [11]
On HRCT, IPF is commonly characterized by patchy and predominantly peripheral, subpleural, and bibasilar reticular opacities. The distribution is predominantly posterior and is often associated with traction bronchiectasis and subpleural honeycombing.
Evidence suggests that alveolar collapse may precede lung fibrosis in IPF, potentially facilitating earlier diagnosis. [25, 26]
Ground-glass attenuations are relatively uncommon; they usually progress to more common reticular attenuations and honeycombing. HRCT scans have been reported to show honeycombing in 90% of patients with IPF. In the absence of honeycombing, the extent of reticular and ground-glass densities can predict a diagnosis of IPF. The probability of IPF exceeds 80% in patients older than 60 years, with one third having reticular densities. [27]
In cases of suspected IPF in which lung HRCT shows more than 30% ground-glass attenuation, consideration should be given to other diagnoses, including desquamative interstitial pneumonitis, idiopathic bronchiolitis obliterans organizing pneumonia, respiratory bronchiolitis–associated interstitial lung disease, hypersensitivity pneumonitis, and nonspecific interstitial pneumonia.
CT detects common complications and associations that may occur with IPF, including acute exacerbation, lung cancer, and dendriform pulmonary ossification, and is useful in informing prognosis based on baseline fibrosis severity. Serial CT imaging is a topic of great interest; this approach may be useful in identifying disease progression before forced vital capacity decline or clinical change is evident. [28]
Pulmonary artery size measured on HRCT has been studied as an outcome predictor in IPF. In a study of 98 IPF patients by Shin et al, the ratio of pulmonary artery diameter to ascending aorta diameter (PA:A) were measured on chest HRCT. Patients with a PA:A ratio >1 had a higher risk of death or transplant compared to patients with a PA:A ratio ≤1 (P< 0.001). [29]
Nuclear Imaging
In cases of IPF, perfusion lung scintigraphy shows nonspecific, subsegmental mismatched perfusion defects. These are not correlated with clinical severity.
Gallium-67 imaging has not proved to be of value in cases of established IPF. [30]
Technetium-99m diethylenetriamine penta-acetic acid (DTPA) is cleared more rapidly when capillary permeability is increased than when it is not, and findings may provide an index of lung inflammation. [31] Fluorodeoxyglucose (FDG) positron emission tomography (PET) may show FDG accumulation in the lung bases; such findings correlate with the honeycomb fibrosis seen on HRCT. [32, 33, 34, 35, 36]
Win et al studied 13 patients with IPF recruited for 2 thoracic FDG-PET studies performed within 2 weeks of each other. For all patients, IPF was diagnosed in consensus at multidisciplinary meetings because of typical clinical, HRCT, and pulmonary function test features. The purpose of this study was to investigate the reproducibility of pulmonary FDG-PET in patients with IPF. Results showed excellent short-term reproducibility in pulmonary FDG uptake among patients with IPF. [37]
Clinical data of 89 IPF patients (mean age: 68.1 yr, male: 94%) who underwent FDG PET/CT for evaluation of lung nodules or cancer staging were retrospectively reviewed by Yoon et al. Mean and maximal standardized uptake values (SUVmean, SUVmax) were measured in the fibrotic area. Adjusted SUV, including SUV ratio (SUVR, defined as SUVmax-to-liver SUVmean ratio), tissue fraction-corrected SUVmean (SUVmeanTF), and SUVR (SUVRTF), and tissue-to-blood ratio (SUVmax/SUVmean venous; TBRblood) were also obtained. The SUVmean, SUVmax, and SUVmeanTF were found to be associated with changes in lung function at 6 months. The SUVR, SUVRTF, and TBRblood were identified as significant predictors for mortality in patients with IPF in the univariate analysis, but not in the multivariate analysis. [38]
Questions & Answers
Overview
What is idiopathic pulmonary fibrosis (IPF)?
What are the diagnostic criteria for idiopathic pulmonary fibrosis (IPF)?
How is the diagnosis of idiopathic pulmonary fibrosis (IPF) confirmed?
Which patient groups are at highest risk for idiopathic pulmonary fibrosis (IPF)?
Which medications are used to treat idiopathic pulmonary fibrosis (IPF)?
How is idiopathic pulmonary fibrosis (IPF) diagnosed?
What is the role of radiography in the workup of idiopathic pulmonary fibrosis (IPF)?
How accurate are radiographic findings in the diagnosis of idiopathic pulmonary fibrosis (IPF)?
What is the role of CT scan in the workup of idiopathic pulmonary fibrosis (IPF)?
What is the role of nuclear imaging in the workup of idiopathic pulmonary fibrosis (IPF)?
-
HRCT of advanced stage of pulmonary fibrosis demonstrating reticular opacities with honeycombing, with predominant subpleural distribution.
-
Bilateral lower lobe opacities and possible mild decrease in lung volumes. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
-
High-resolution CT (HRCT) shows increased pulmonary attenuation with distortion of the pulmonary architecture. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
-
High-resolution CT (HRCT) shows distortion of the pulmonary architecture with thickening of pulmonary interstitium and some areas of ground-glass attenuation. No obvious honeycombing is present. Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
-
Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.
-
Courtesy of Sat Sharma, MD, FRCPC, FACP, FCCP, DABSM.