Practice Essentials
Achondroplasia is an inherited disorder of bone growth that causes the most common type of dwarfism [1] and belongs to one of the groups of disorders collectively called chondrodystrophies. Achondroplasia is the most common form of disproportionate short stature and occurs in 1 in 20,000 live births. It is associated with potentially serious complications such as foramen magnum and spinal stenosis, which result in increased morbidity and mortality. The disorder is caused by a dominantly inherited FGFR3 mutation that permanently activates the fibroblast growth factor receptor 3 (FGFR3) and its downstream mitogen-activated protein kinase (MAPK) signaling pathway. This inhibits chondrocyte differentiation, and growth plate function is severely compromised. [2, 3, 4]
Achondroplasia can be diagnosed on the basis of characteristic clinical and radiographic findings in most affected individuals. Abnormal bone growth results in short stature with disproportionately short arms and legs, [5] a large head with frontal bossing, a narrow thorax, a waddling gait, and characteristic facial features. Intelligence and life span are usually normal, though the risk of infant death from compression of the cervical spinal cord and/or upper airway obstruction is increased. [6]
Although more than 100 skeletal dysplasias that cause short stature are recognized, many are extremely rare, and all have clinical and radiographic features that readily distinguish them from achondroplasia. Results on antenatal ultrasonograms either suggest or confirm most skeletal dysplasias. Conditions that may be confused with achondroplasia include achondrogenesis; chondroectodermal dysplasia; asphyxiating thoracic dystrophy; osteogenesis imperfecta; congenital hypophosphatasia; metatrophic dysplasia; Roberts syndrome; diastrophic dysplasia; short rib-polydactyly syndrome types I, II, and III; spondyloepiphyseal dysplasia congenita (camptomelic dysplasia); thanatophoric dysplasia; fibrochondrogenesis; chondrodysplasia punctate (rhizomelic type); Kniest dysplasia; mesomelic and acromesomelic dysplasia; hypochondroplasia; pseudoachondroplasia; and double heterozygosity in bone growth disorders.
(See the images below.)
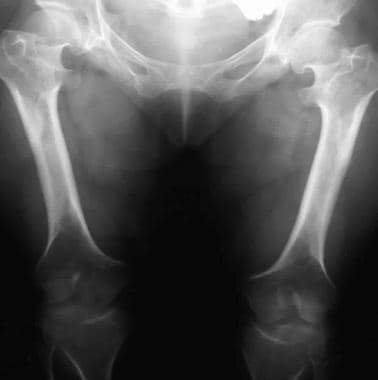 Genu varum. Image shows rhizomelic shortening of the bilateral femurs with metaphyseal flaring. The bones are wide because of unaffected appositional growth.
Genu varum. Image shows rhizomelic shortening of the bilateral femurs with metaphyseal flaring. The bones are wide because of unaffected appositional growth.
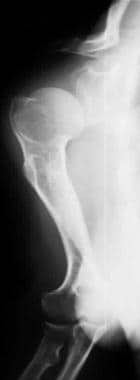 Image shows rhizomelic shortening of the humerus with posterior bowing and an incomplete glenoid fossa.
Image shows rhizomelic shortening of the humerus with posterior bowing and an incomplete glenoid fossa.
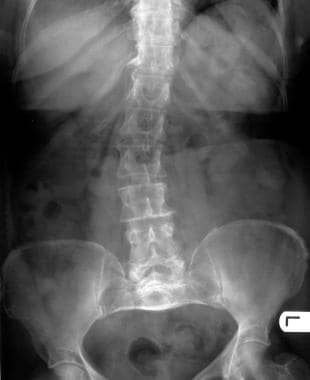 The spine is often affected in achondroplasia. Features include interpediculate narrowing and thickened pedicles.
The spine is often affected in achondroplasia. Features include interpediculate narrowing and thickened pedicles.
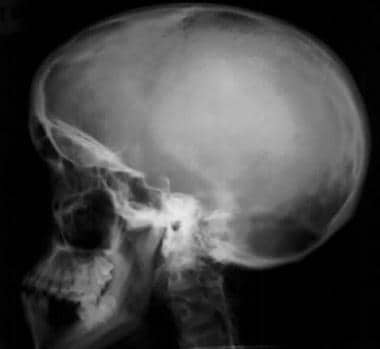
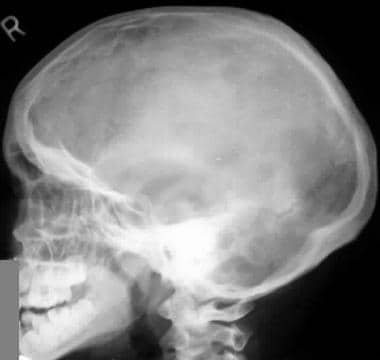 Image shows an enlarged calvaria with a shortened skull base and frontal bossing. Note the midface hypoplasia.
Image shows an enlarged calvaria with a shortened skull base and frontal bossing. Note the midface hypoplasia.
Skeletal anomalies associated with achondroplasia reflect retarded endocardial bone formation. Therefore, the long bones are short but wide because appositional bone growth is unaffected. [7] The skull is not dependent on endocardial bone; therefore, it is generally large. The spinal column is of relatively normal length but becomes kyphotic as a result of vertebral anomalies and body habitus. The achondroplastic foramen magnum is small at birth. This markedly diminished growth results not only from abnormal endochondral bone growth but also from abnormal placement and premature fusion of the synchondroses.
In people with achondroplastic dwarfism, stenosis of the spinal canal is secondary to abnormalities of endochondral ossification with premature synostosis of the ossification centers of the vertebral body and the posterior arch. This results in thickening of the laminae, shortening of the pedicles, and a reduction in the height of the vertebral bodies. Additional factors, such as prolapsed intervertebral disks, osteophytes, and progressive thoracolumbar kyphosis, contribute to the narrowing of the spinal canal. [8]
Preferred examination
Prenatal diagnosis of achondroplasia can be achieved with ultrasonography. Antenatal, targeted ultrasonography is indicated in at-risk pregnancies. Conventional radiography remains the preferred modality for initial investigation for both children and adults. Typically, a complete skeletal survey (or a hemi-survey of one side of the body) will be obtained. However, most of the diagnostically critical features will be appreciated on a single anteroposterior radiograph of the pelvis and femora. [9]
Myelography, computed tomography (CT) scanning, CT myelography, and magnetic resonance imaging (MRI) are added when indicated, as with compressive cord symptoms at the craniocervical and thoracolumbar junctions. Both CT scanning and MRI can be used to examine the size of the foramen magnum, which is an important determinant of compressive myelopathy of the upper cervical cord. MRI has the added advantage of depicting posterior cranial fossa anatomy and other abnormalities, such as syringomyelia and hydrocephalus.
MRI is the modality of choice in cases of suspected spinal stenosis; disk lesions; and compromise of the exiting nerve root in the lumbar region. A cranial MRI is recommended to identify spinal cord compression due to foramen magnum stenosis and to identify ventricular enlargement with neurological symptoms (hydrocephalus). [3]
Ultrasonography provides a noninvasive and fairly reliable method of assessing the ventricles of infants before the fontanels close. Ultrasonography may be supplemented with CT scanning and/or MRI of the head to monitor for compression of the foramen magnum. [10]
Limitations of techniques
Both false-positive and false-negative diagnoses may occur with antenatal ultrasonography of skeletal dysplasias. Heterozygous disease may not be recognized until late in the second trimester (>24-28 wk), because ultrasonograms are normal early in the course of the disease.
Ultrasonography remains operator dependent. Conventional radiography is good for depicting skeletal pathology, but it is poor at providing information on the brain and spinal cord.
Myelography is invasive; however, this technique has traditionally been the most useful examination in delineating the level of compression of the spinal cord or the cauda equina. However, in patients with achondroplasia, myelography is difficult because of stenosis of the lumber spine. When myelography is performed near the lumber root, there is a risk that the neurologic deficit will increase; this is particularly so in cases of kyphosis. Therefore, when myelography is necessary, the study is best performed by means of cisternal puncture; however, this step makes the procedure even more invasive than it otherwise would be.
CT scanning exposes the patient to ionizing radiation and is limited in its capacity to portray the spinal cord and to depict structures of the posterior fossa. With this modality, infants and young children may need sedation or general anesthesia.
MRI is frequently used to evaluate the brain and spinal cord in patients with achondroplasia. This modality can accurately depict anatomic encroachment on the central nervous system (CNS). However, MRI is expensive and creates problems for patients with claustrophobia, and it cannot be performed in patients with cardiac pacemakers or in patients who have certain surgical clips or other foreign objects in the body. As with CT scanning, young children may need sedation or general anesthesia to undergo MRI.
In the past, isotopic cisternography with an intrathecal injection of indium-111 (111In) diethylenetriamine pentaacetic acid (DTPA) was commonly used to assess hydrocephalus. This technique is rarely used now, because it does not increase accuracy in predicting favorable responses to shunting, relative to CT scanning and clinical evaluation.
Radiography
Conventional radiography is noninvasive, inexpensive, quick to perform, and fairly reliable. This imaging modality is generally the first examination performed after birth to confirm the diagnosis of achondroplasia. It is also usually the first examination of choice for patients with a symptomatic spine. Conventional radiographs generally provide only bony detail and provide little information about the state of the brain and spinal cord. The cord is better depicted with myelography, although myelography provides little information about anomalies within the cord.
The radiographic findings in achondroplasia are as follows:
-
Shortening of tubular bones with a normal shaft caliber
-
Short extremities and ribs-versus-trunk length (see the following image)
-
Short phalanges
-
Ball-in-socket epiphyses
-
Metaphyseal flaring and cupping
-
Circumflex or chevron seat on the metaphysis
-
Squared iliac wings and narrow sacroiliac notch (champagne glass) (see the images below)
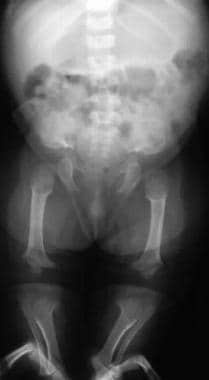 Image shows progressive narrowing of the interpediculate distance with a champagne-glass pelvis. Note that the legs are straight in infancy.
Image shows progressive narrowing of the interpediculate distance with a champagne-glass pelvis. Note that the legs are straight in infancy.
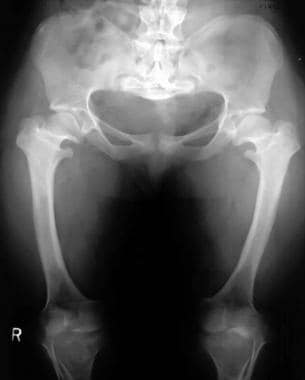 Champagne-glass pelvis with squared iliac wings, a narrow sacroiliac notch, and a reduced acetabular angle.
Champagne-glass pelvis with squared iliac wings, a narrow sacroiliac notch, and a reduced acetabular angle.
-
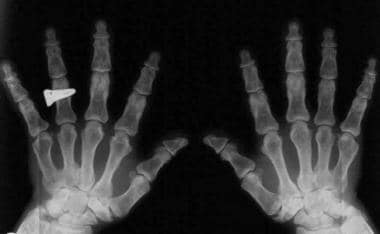
Fingers widely opposed and of equal length (trident hands) (see the following images)
-
Enlarged skull vault and mandible (see the images below)
 Image shows an enlarged calvaria with a shortened skull base and frontal bossing. Note the midface hypoplasia.
Image shows an enlarged calvaria with a shortened skull base and frontal bossing. Note the midface hypoplasia.
-
Small foramen magnum
-
Narrow anteroposterior (AP) diameter of the spine with a concave posterior surface (see the following image)
-
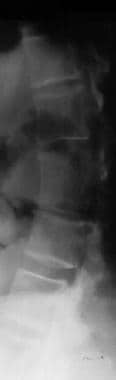
Decreased lumber interpediculate distance and narrow spinal canal; narrow vertebral interpediculate distance (see the images below)
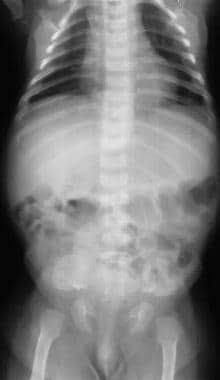 Image shows progressive reduction in vertebral interpediculate distance in the caudal direction.
Image shows progressive narrowing of the interpediculate distance with a champagne-glass pelvis. Note that the legs are straight in infancy.
Image shows progressive reduction in vertebral interpediculate distance in the caudal direction.
Image shows progressive narrowing of the interpediculate distance with a champagne-glass pelvis. Note that the legs are straight in infancy.
-
Hypoplastic (bullet nose) thoracolumbar junction vertebrae (see the following images)
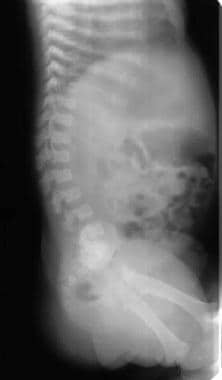 Image shows progressive narrowing of the lumbar spinal canal, bullet-nose vertebrae, and marked lumbar lordosis. Note the shortened ribs.
Image shows progressive narrowing of the lumbar spinal canal, bullet-nose vertebrae, and marked lumbar lordosis. Note the shortened ribs.
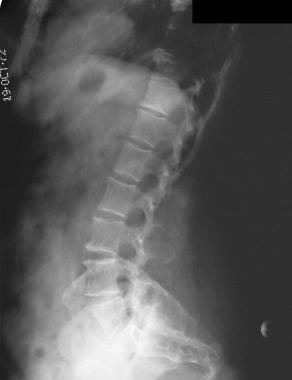 Anterior wedging of the vertebral bodies produces a bullet shape (not shown). Disk herniation is common. Changes in the spine can result in stenosis of the spinal canal, particularly in the lumbar region.
Anterior wedging of the vertebral bodies produces a bullet shape (not shown). Disk herniation is common. Changes in the spine can result in stenosis of the spinal canal, particularly in the lumbar region.
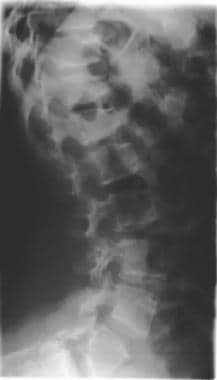 Image shows bullet-nose hypoplastic vertebrae with a narrowed anteroposterior diameter of the spine.
Image shows bullet-nose hypoplastic vertebrae with a narrowed anteroposterior diameter of the spine.







Kitoh and associates found that posterior bowing of the distal humerus in patients with achondroplasia is a principal cause of loss of extension of the elbow, and posterior dislocation of the radial head causes further limitation of movement in the more severely affected joints. [11] The investigators examined extension of the elbow clinically, and measured the angle of posterior bowing of the distal humerus on lateral radiographs. Extension of the elbow was limited in 68% of the elbows in the study, with a mean loss of extension of 13.1°; posterior bowing of the humerus was seen in all elbows, with a mean angle of 17.0°.
The investigators noted that posterior bowing greater than 20° caused a loss of full elbow extension, [11] Twenty-two percent of the elbows demonstrated posterior dislocation of the radial head. The mean loss of extension of the elbows was 28.7°; this loss of extension was significantly greater than the loss seen in elbows in which the head was not dislocated (8.7°), although posterior bowing was not significantly different between the groups (19.3° vs 16.3°). [11]
Thomeer and van Dijk found that dynamic lumbar myelography was required for demonstrating the symptomatic level in the surgical treatment of lumbar stenosis in patients with achondroplasia that involved selective widening of the lumbar interapophyseolaminar diameter. [12]
Computed Tomography
The sensitivity of CT myelography is greater than that of conventional myelography. CT scanning depicts bone detail better than MRI. Although MRI has an obvious advantage of being radiation free, many clinicians believe that the degree of stenosis is usually best demonstrated with myelography.
Details of the posterior fossa brain and cord are better depicted on MRI than on CT studies. Cord edema and changes associated with myelomalacia usually cannot be seen with CT scanning. CT scans also provide only indirect evidence of associated anomalies, such as syringomyelia, whereas MRI shows such features directly and clearly.
Among infants with achondroplasia and apnea, CT scanning and MRI have repeatedly demonstrated cord compression resulting from direct impingement of the posterior rim of the foramen magnum and C1 arch. Sleep apnea responds well to decompression of the foramen magnum. In addition, CT scanning shows that in virtually all children with achondroplasia, there is some degree of compromise of the foramen magnum. In about 96% of such children, the foramen magnum is smaller than 3 standard deviations of the mean; both CT scanning and/or MRI can depict this change.
A small spinal canal is present in the cervical region from birth, but symptoms of cervical canal stenosis generally do not occur until middle age or later. If neurologic deficit occurs and does not resolve through conservative measures, laminectomy at multiple levels may be required. Preoperative imaging with CT scanning, CT myelography, and/or MRI is vital for successful surgery. Preoperative and intraoperative myelography, CT evaluation, or MRI defines the level of cord and/or root compression caused by dorsolumbar spinal stenosis. Although MRI has largely replaced conventional myelography, CT myelography and intraoperative myelography may still play a role.
Otitis media is a relatively common complication of achondroplasia that can result in changes affecting the temporal bone that might predispose patients with achondroplastic dwarfism to otitis media. With high-resolution CT examination of the temporal bone, Cobb et al found the most notable change was the rotational effect, which was most pronounced medially and which resulted in abnormal orientations of inner-ear structures relative to middle-ear structures and of middle-ear structures relative to the external auditory canal. [13] There were also a number of morphologic changes: (1) poor development of mastoid air cells, (2) shortening of the carotid canals, (3) narrowing of the skull base, (4) towering petrous ridges, and (5) relative rotation of the cochlea and other structures of the temporal bone. [13]
The investigators also noted a lack of evidence of otitis media or its sequelae in any of the patients with achondroplasia. In addition, audiogram results showed evidence of mixed hearing loss in the 4 children but only of sensorineural hearing loss in the adults. [13] The authors concluded that the persistent hearing loss in patients with achondroplasia is not a sequela of otitis media, as other authors have suggested. Intrinsic vestibulocochlear changes below the limits of resolution of high-resolution CT scanning may be responsible. [13]
Magnetic Resonance Imaging
MRI is a noninvasive technique and is ideal for children because it does not use ionizing radiation. In addition, MRI has an advantage over CT scanning in the degree of detail of the posterior cranial fossa cord that it provides. Craniocervical MRI findings may include narrowing of the foramen magnum and C1 canal, effacement of the subarachnoid spaces at the cervicomedullary junction, abnormal intrinsic cord signal intensity, and mild to moderate ventriculomegaly. In the spinal canal, associated anomalies such as syringomyelia and changes of myelomalacia are well depicted on MRI. In addition to depicting spinal canal stenosis, MRI also demonstrates disk protrusions and osteophytes that cause compromise (see the image below). [14, 15, 3]
Early clinical and MRI evaluations are necessary to determine whether infants with achondroplasia have cervicomedullary compression. With early recognition, an immediate decompression can be performed safely to avoid serious complications associated with cervicomedullary compression, including sudden death. [16] CT scanning depicts details of bone and the degree of spinal stenosis better than MRI. Claustrophobia may limit the quality of MRI, and motion artifacts may produce false-negative and/or false-positive results.
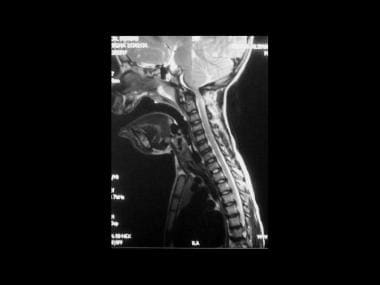 Achondroplasia. Sagittal section of the cervical spine on a T2-weighted magnetic resonance image in a 6-year-old patient who presented with a neurologic deficit. This image shows narrowing of the foramen magnum at the C1 canal, effacement of the subarachnoid spaces at the cervicomedullary junction, and abnormal intrinsic cord signal intensity.
Achondroplasia. Sagittal section of the cervical spine on a T2-weighted magnetic resonance image in a 6-year-old patient who presented with a neurologic deficit. This image shows narrowing of the foramen magnum at the C1 canal, effacement of the subarachnoid spaces at the cervicomedullary junction, and abnormal intrinsic cord signal intensity.
Bruhl et al found an unexpectedly poor correlation between MRI and clinical findings in achondroplastic children with regard to indications for decompressive surgery at the craniocervical junction (CCJ) and suggested that the role of MRI is limited to demonstrating spinal cord compression in individual cases. [17]
In a study of atlantoaxial subluxation using MRI, Yamashita et al demonstrated that the severity of myelopathy had a poor correlation with the atlantodental interval on conventional radiography; however, there was a high correlation between magnetic resonance grading and the degree of myelopathy. [18]
Kao et al suggested that MRI is useful in delineating the many abnormalities of the cranial, cerebral, and cervicomedullary junction present in children with achondroplasia. [19] All 10 children in their study had narrowing of the subarachnoid space at the level of the foramen magnum, and 5 had compressive deformities of the cervicomedullary junction. In addition, apparent upward displacement of the brainstem and a relatively vertical course of the optic nerve were seen in all patients; dilated lateral and third ventricles were seen in 5 patients; bifrontal widening of the subarachnoid space was evident in 4 children; skull asymmetry was seen in 2 patients; and an empty sella (confirmed on metrizamide cisternography) was present in 1 child. In 1 patient, foci of abnormal signal intensity were seen in the cervicomedullary region. [19]
The amount of blood flow in the superior sagittal sinus is correlated with brain maturation. Hydrocephalus associated with achondroplasia is closely related to reduction in blood flow in the superior sagittal sinus—a finding that supports the hypothesis that in patients with achondroplasia, hydrocephalus results from a restriction of venous outflow.
Cine phase-contrast MRI is a convenient and effective method of measuring volumetric flow rates in vivo. In healthy children, the flow velocity is 92-196 mm/s (mean, 136 mm/s), and the flow rate is 189-688 mL/min (mean, 484 mL/min). The flow rates change in a manner that is statistically related to age: flow rate rapidly increases during the first 2 years and reaches a peak by 6-8 years of age. The flow velocity shows a similar pattern but without significant statistical correlation. In all cases of achondroplasia with hydrocephalus, both flow values are lower than the reference values by 1 standard deviation. In cases of achondroplasia without hydrocephalus, as well as in obstructive hydrocephalus, the values are not reduced. [20, 21, 22]
High-signal intramedullary cord lesions in achondroplasia on T2-weighted MRI just below the craniocervical junction are a common finding. These lesions are usually asymptomatic and unaccompanied by cord compression signs. The lesions occur in a third of the achondroplasia population. Their etiology is unclear and seems to be unrelated to mechanical causes. [23]
Ultrasonography
Ultrasonography is generally performed in the antenatal setting and in pregnant women whose fetuses are at risk for achondroplasia. Serial ultrasonography enables prenatal distinction between homozygous and heterozygous disease. [24, 25] However, approximately 80% of cases are due to a de novo mutation, and most cases of achondroplasia occur in low‐risk pregnancies. Identification of short fetal limbs during the late second trimester or third trimester is the cornerstone of the diagnosis of achondroplasia. [26]
Homozygous achondroplasia results in rhizomelic micromelia, normal trunk length, and cloverleaf skull; these cases are lethal. Lung hypoplasia is a major cause of mortality (associated with thoracic narrowing). [27] There is a noticeable disproportion between skull dimensions and/or biparietal diameter (BPD) and limb lengths. The discrepancy between femoral length and BPD is noted as early as 13 weeks' gestation. The femoral length decreases to below the third percentile at 14.0-16.5 weeks' BPD age (mean, 15.6 wk). Therefore, femoral growth curves are established in the second trimester.
Heterozygous disease may not be recognized until late in the second trimester (>24-28 wk); early in the course of disease, ultrasonograms are normal. The changes in heterozygous achondroplasia are relatively mild and include short limbs, narrow thorax and abdomen, increased fetal head circumference and BPD, a protuberant forehead, and a diminished interpediculate distance in the spine. Rhizomelic limb shortening that predominantly affects the proximal long bones is observed.
Krakow and associates found that 3-dimensional (3-D) imaging in the prenatal-onset diagnosis of skeletal dysplasia had advantages over 2-dimensional (2-D) imaging in the evaluation of facial dysmorphism, relative proportion of the appendicular skeletal elements, and the hands and feet. [28] Of most importance, the patient and referring physician appreciated the 3-D images of the abnormal findings more readily than other images; this advantage aided in patient counseling and in managing the pregnancy. [29]
Patel and Filly retrospectively reviewed serial sonograms of 15 fetuses at 25% risk of homozygous achondroplasia. [30] Femoral growth curves were established and were compared with published standards to determine the gestational age. They were calculated according to BPD, at which femoral length crossed below the third percentile. The presence and severity of achondroplasia were clinically determined after birth. Their results showed that the femoral length crossed the third percentile at 14.0-16.5 weeks BPD age (mean, 15.6 wk) in the 4 homozygous fetuses and at 18.2-26.2 weeks BPD age (mean, 21.5 wk) in the 8 heterozygous fetuses. In the 3 unaffected fetuses, femoral length did not cross percentiles as gestational age increased.
Parilla and associates reported that the use of serial ultrasonographic scans to establish a femoral growth curve in the second trimester enables prenatal distinction between homozygous, heterozygous, and unaffected fetuses when both parents have heterozygous achondroplasia. The investigators reviewed 37 cases of skeletal dysplasia diagnosed antenatally over 8 years, in which complete follow-up was available in 31 cases. The mean gestational age at diagnosis was 22.7 weeks (range, 14-32.3 wk). In 21 cases, the diagnosis was made before the 24th week. A final diagnosis was obtained in 80% of cases. The antenatal diagnosis was correct in 20 (65%) of 31 cases. Two false-positive diagnoses occurred. [31]
In the study by Parilla et al, specific final diagnoses included thanatophoric dysplasia (N=8), osteogenesis imperfecta (N=6), Roberts syndrome (N=2), achondroplasia (N=3), Ellis-van Creveld syndrome (N=1), metaphyseal dysplasia (N=1), spondyloepiphyseal dysplasia (N=1), distal arthrogryposis (N=1), caudal regression (N=1), and glycogen storage disorder (N=1). The condition was correctly thought to be lethal in 16 of the fetuses on the basis of early, severe long-bone shortening (N=13), femur length–abdominal circumference ratio of less than 0.16 (N=12), hypoplastic thorax (N=10), marked bowing or fractures (N=4), short ribs (N=4), caudal regression (N=1), and cloverleaf skull (N=1). The ability to predict lethality was 100%. No false-positive findings with respect to lethality occurred. [31]
Parilla et al concluded that antenatal diagnosis of skeletal dysplasias is problematic: In their series, only 20 of 31 cases were correctly diagnosed. However, the antenatal prediction of lethality was highly accurate. The most common predictors of lethal skeletal dysplasias included early and severe shortening of the long bones, femur length–abdominal circumference ratio of less than 0.16, hypoplastic thorax, and certain distinguishing characteristics. [31]
-
Genu varum. Image shows rhizomelic shortening of the bilateral femurs with metaphyseal flaring. The bones are wide because of unaffected appositional growth.
-
Image shows rhizomelic shortening of the humerus with posterior bowing and an incomplete glenoid fossa.
-
Image shows inverted femoral physes (inverted V configuration), which contributes to a waddling gait.
-
Image shows progressive narrowing of the lumbar spinal canal, bullet-nose vertebrae, and marked lumbar lordosis. Note the shortened ribs.
-
Image shows progressive reduction in vertebral interpediculate distance in the caudal direction.
-
The spine is often affected in achondroplasia. Features include interpediculate narrowing and thickened pedicles.
-
Anterior wedging of the vertebral bodies produces a bullet shape (not shown). Disk herniation is common. Changes in the spine can result in stenosis of the spinal canal, particularly in the lumbar region.
-
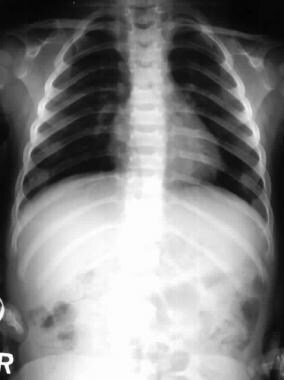
Shortened ribs.
-
Image shows progressive narrowing of the interpediculate distance with a champagne-glass pelvis. Note that the legs are straight in infancy.
-
Image shows an enlarged calvaria with a shortened skull base and frontal bossing. Note the midface hypoplasia.
-
Enlarged calvaria. Note the enlarged mandible.
-
Image shows posterior bowing of the humerus, the principal cause of the loss of elbow extension. Posterior dislocation of the radial head may also contribute.
-
Champagne-glass pelvis with squared iliac wings, a narrow sacroiliac notch, and a reduced acetabular angle.
-
Patient with achondroplasia and less severe pelvic changes.
-
Trident hands. Image shows widely opposed fingers of equal length.
-
Trident hands.
-
Image shows a decreased lumbar interpedicular distance. Note the scoliosis.
-
Image shows concave scalloping of the posterior surface of the vertebral bodies.
-
Image shows scoliosis.
-
Image shows bullet-nose hypoplastic vertebrae with a narrowed anteroposterior diameter of the spine.
-
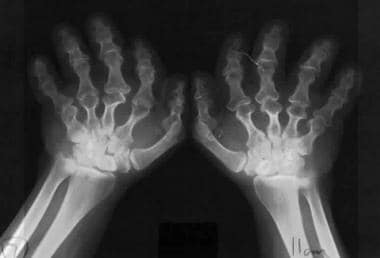
Ellis-van Creveld (EVC) syndrome is a differential diagnosis of short-limb dwarfisms. It is also known as chondroectodermal dysplasia. This autosomal recessive disease involves chromosome 4p16. The hands demonstrate polydactyly in almost all patients, whereas the feet demonstrate polydactyly in only 10%. Note the broad hands with short middle phalanges and hypoplastic distal phalanges. The carpal bones are malformed, with fusion of the capitate and hamate. Extracarpal bones might also be present. The ends of the ulna and radius are enlarged.
-
Ellis-van Creveld (EVC) syndrome. (See the previous image.)
-
The knees of patients with Ellis-van Creveld (EVC) syndrome develop a genu valgus deformity, and the long bones are short. Hypoplasia of the proximal tibia is also present. (See the previous 2 images.)
-
The knees of patients with Ellis-van Creveld (EVC) syndrome develop a genu valgus deformity, and the long bones are short. Hypoplasia of the proximal tibia is also present. (See the previous 3 images.)
-
The thoracic cavity of this patient with Ellis-van Creveld (EVC) syndrome is small and narrow, with short ribs. About 60% of patients have cardiac anomalies, and most patients ultimately die from respiratory illness.
-
In Ellis-van Creveld (EVC) syndrome, the teeth are hypoplastic, as are the nails. The teeth are small and cone shaped, with irregular spacing. Other facial anomalies include a partial harelip.
-
Metaphyseal chondroplasia (Schmid type) is a differential diagnosis of achondroplasia, with metaphyseal flaring of the ulna and radius as well as bowing of the shaft. Note no hand involvement with metaphyseal chondroplasia, unlike achondroplasia.
-
Metatrophic dwarfism II, or Kniest syndrome, is a differential diagnosis. Skeletal dysplasia results in short limbs and a proportionally long trunk; however, the head and face appear normal. With time, severe kyphoscoliosis produces marked shortening of the trunk, which can make body proportions deceiving.
-
Achondroplasia. Sagittal section of the cervical spine on a T2-weighted magnetic resonance image in a 6-year-old patient who presented with a neurologic deficit. This image shows narrowing of the foramen magnum at the C1 canal, effacement of the subarachnoid spaces at the cervicomedullary junction, and abnormal intrinsic cord signal intensity.








