Practice Essentials
Thalassemic syndromes are genetically determined disorders of hemoglobin synthesis with decreased production of either alpha or beta polypeptide chains of hemoglobin (Hb) molecules. This reduced production results from markedly decreased amounts of globin messenger ribonucleic acid. The imbalance may result from many genetic lesions. Although there are many types of thalassemic syndromes, each involves decreased production of one globin chain or more, which form the different Hbs normally found in RBCs. In clinical practice, the most important types affect either α- or β-chain synthesis.
Although routine transfusion has increased the life expectancy associated with severe thalassemias, complications of chronic blood exposure are now the predominant challenge in disease management. Imaging is used primarily to monitor iron chelation therapy. Scientific studies have led to better understanding of iron trafficking, including how iron accumulates in tissues that were not intended to store iron. These studies have elucidated the need to monitor not only serum parameters but also hepatic iron and even iron in other tissues. [1] Noninvasive magnetic resonance imaging (MRI) has replaced invasive liver biopsy as the preferred modality to measure liver, cardiac, pancreatic, and even pituitary iron. [2]
The Thalassaemia International Federation (TIF) recommends that assessment of iron overload status through liver iron concentration (LIC) measurement be done by MRI evaluation. Other liver iron concentration measurement techniques may be used when MRI is unavailable. Assessment of LIC should be done at baseline and every 12-24 months in patients who are not receiving iron chelation therapy and every 6-12 months (or earlier as needed for iron chelator dose modification) in patients receiving iron chelation therapy. [3]
Cardiovascular magnetic resonance (CMR) T2* is the method of choice for the assessment of cardiac iron, and in the UK, where it was first introduced clinically, 60% reductions in overall mortality for thalassemia major have been observed. [4] CMR provides a more direct assessment of cardiac iron, and as T2* sequences have become more widely available, MRI has become the standard for monitoring cardiac iron loading. [5]
CT scanning is rarely of value, yet it may define the anatomy of change in complex bones and confirm the diagnosis of extramedullary hematopoiesis. [6] Although CT scanning confirms the deposition of iron in the liver and other organs, correlation with organ damage, serum ferritin levels, and transfusional history is unsatisfactory.
(See the images below.)
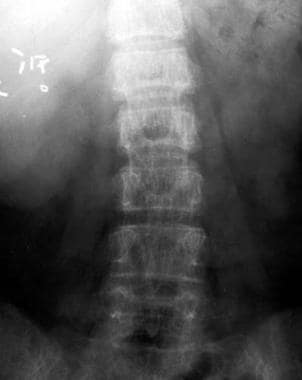 Thalassemia. Anteroposterior radiograph of the lumbar spine. Osteopenia is present; note striated appearance of the vertebral bodies resulting from preservation and thickening of the vertical trabeculae. Clips from a previous cholecystectomy are present.
Thalassemia. Anteroposterior radiograph of the lumbar spine. Osteopenia is present; note striated appearance of the vertebral bodies resulting from preservation and thickening of the vertical trabeculae. Clips from a previous cholecystectomy are present.
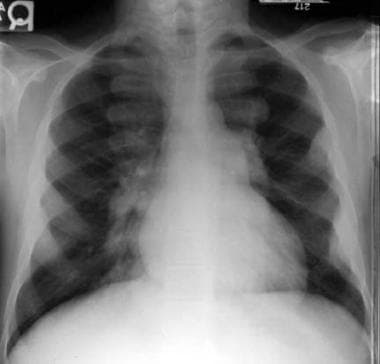 Thalassemia. Extramedullary hematopoiesis. Lobulated soft tissue opacities are noted overlying the ribs anteriorly and posteriorly.
Thalassemia. Extramedullary hematopoiesis. Lobulated soft tissue opacities are noted overlying the ribs anteriorly and posteriorly.
The most common forms of α-thalassemia are as follows:
-
Silent carrier α-thalassemia: The diagnosis cannot be confirmed based on Hb electrophoresis results, which are usually normal in all α-thalassemia traits
-
α-Thalassemia trait: Characterized by mild anemia and low RBC indices
-
Hb H disease: Represents α-thalassemia intermedia, with mildly to moderately severe anemia, splenomegaly, icterus, and abnormal RBC indices
-
α-Thalassemia major: Results in the severe form of homozygous α-thalassemia
Some of the more common forms of β-thalassemia are as follows:
-
Silent carrier β-thalassemia: Patients are asymptomatic, except for possible low RBC indices
-
β-Thalassemia trait: Patients have mild anemia, abnormal RBC indices, and abnormal Hb electrophoresis results with elevated levels of Hb A2, Hb F, or both
-
Thalassemia intermedia: Patients have anemia of intermediate severity
-
Β-thalassemia associated with β-chain structural variants: The most significant condition in this group of thalassemic syndromes is the Hb E/β thalassemia
-
Thalassemia major (Cooley anemia): This condition is characterized by transfusion-dependent anemia, massive splenomegaly, bone deformities, growth retardation, and peculiar facies in untreated individuals, 80% of whom die within the first 5 years of life from complications of anemia
On the basis of clinical severity and transfusion requirement, thalassaemia syndromes can be classified phenotypically into 2 main groups: transfusion-dependent thalassemias (TDTs) and non-transfusion-dependent thalassemias (NTDTs). The Thalassaemia International Federation (TIF) has released separate guidelines for the diagnosis and management of TDTs and NTDTs. [7, 3]
Thalassemia is a laboratory diagnosis. In areas with a high incidence of thalassemia, universal screening of neonates is recommended for both α- and β-thalassemia disorders. In addition, advances in capillary electrophoresis and molecular testing have improved the specificity and availability of diagnosis outside the neonatal period. [7]
Radiography
Plain radiographs are not usually required to confirm the diagnosis of thalassemai. However, when radiographs are utilized, the complete skeleton is involved. [8, 9] Radiographically, the skeletal response to marrow proliferation consists of expansion of the medulla, thinning of cortical bone, and resorption of cancellous bone, which results in a generalized loss of bone density. [10, 11] Frequently, small areas of lucency resulting from focal proliferation of marrow may be present, often demarcated by coarsened but less numerous trabeculae. In addition, the hypertrophic and hyperplastic marrow may perforate the cortex, proliferate subperiosteally, and stimulate a variety of periosteal responses. Depending on the bone, these factors may result in a variety of radiographic appearances.
Vertebral bodies
In weight-bearing bones, the resorptive process preserves the primary trabeculae at the expense of secondary trabeculae. In the vertebral bodies, this results in a striated appearance resulting from thickened vertical trabeculae that stand out against the paucity of horizontal trabeculae (see the image below). In severely affected patients, biconcavity of the superior and inferior margins of the vertebral bodies or compression fractures may occur.
Thalassemia. Anteroposterior radiograph of the lumbar spine. Osteopenia is present; note striated appearance of the vertebral bodies resulting from preservation and thickening of the vertical trabeculae. Clips from a previous cholecystectomy are present.
Skull and facial bones
In severely affected patients, a widening of the diploic space (medulla) with a thinning of the tables (cortices) occurs, frequently with complete obliteration of the outer table. New bone forms in response to marrow proliferation beneath the periosteum. These bony spicules may be seen radiographically and result in a classic "hair-on-end" appearance. [12] Because it lacks hematopoietic marrow, the occipital bone usually is not involved. (See the image below.)
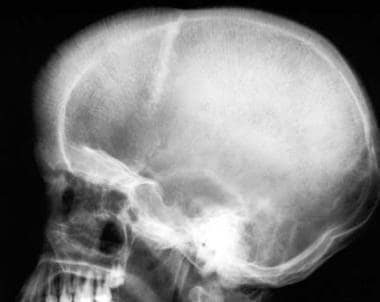 Thalassemia. Lateral skull radiograph. Widening of the calvarium is present; the outer table overlying the frontal region is obliterated and new bone formation can be seen in the diploë, producing a "hair-on-end" appearance. Note that the occipital bone is uninvolved. The impressions of the calvarial vessels are enlarged.
Thalassemia. Lateral skull radiograph. Widening of the calvarium is present; the outer table overlying the frontal region is obliterated and new bone formation can be seen in the diploë, producing a "hair-on-end" appearance. Note that the occipital bone is uninvolved. The impressions of the calvarial vessels are enlarged.
Proliferation of marrow within the frontal and facial bones impedes pneumatization of the paranasal sinuses. This results in hypertrophy of osseous structures and a consequent prominence of the lateral margins of the malar eminences, together with anterior and medial displacement of developing teeth. These features explain the clinical findings noted by Cooley. Characteristically, ethmoidal sinuses are not involved, a factor attributable to the absence of red marrow in the sinus walls.
Appendicular skeleton
In patients with more profound anemia, changes may be noted in the distal bones of the extremities. In the most severely affected patients, the phalanges reveal the above-noted changes of cortical thinning, osteopenia, and coarsening of the trabeculae and may lose their normal tubulation, which frequently results in a squared or sausage-shaped configuration.
(See the image below.)
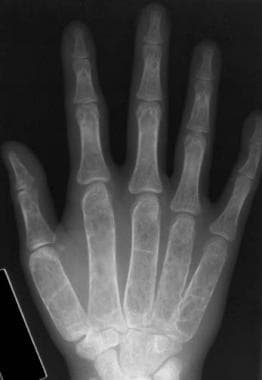 Thalassemia. Anteroposterior of the hands. A generalized loss of bone density is observed. The cortex is thinned, and the trabeculae are coarsened and outline localized lucency. Widening of the medullary cavity has resulted in squaring of the metacarpals. Note the enlarged nutrient foramina in the middle phalanges.
Thalassemia. Anteroposterior of the hands. A generalized loss of bone density is observed. The cortex is thinned, and the trabeculae are coarsened and outline localized lucency. Widening of the medullary cavity has resulted in squaring of the metacarpals. Note the enlarged nutrient foramina in the middle phalanges.
Fractures may occur, although less commonly than expected from the degree of osteoporosis (see the image below). The sedentary life these children are forced to live may possibly protect them from frequent injury. In severely affected patients, well-defined erosions of the periosteal margin of the cortex of the metaphysis or diaphysis may be identified.
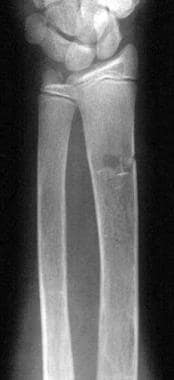 Thalassemia. Anteroposterior radiograph of the forearm. A fracture is noted in the distal radius. Evidence of medullary expansion and cortical thinning is observed.
Thalassemia. Anteroposterior radiograph of the forearm. A fracture is noted in the distal radius. Evidence of medullary expansion and cortical thinning is observed.
Ribs
Similarly to the long bones, the ribs may reveal a spectrum of radiographic findings that demonstrate the varied response of the medulla, cortex, and periosteum to proliferating marrow. Evidence of widening, generalized osteopenia, or localized lucency resulting from hypertrophy and hyperplasia of marrow within the medulla may be seen. Cortical erosions may be pronounced and are considered to result from focal subperiosteal proliferation of marrow.
(See the image below.)
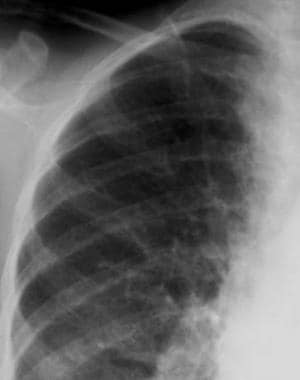 Thalassemia. Radiograph of the ribs. Erosions of the inferior cortical margins of the third, fourth, and fifth ribs are noted.
Thalassemia. Radiograph of the ribs. Erosions of the inferior cortical margins of the third, fourth, and fifth ribs are noted.
A frequent finding has been referred to as the "rib-within-a-rib" appearance and is noted particularly in the middle and anterior portions of the ribs. The finding consists of a long linear density within or overlapping the medullary space of the rib and running parallel to its long axis (see the image below). This appearance is not noted in any other portion of the skeleton.
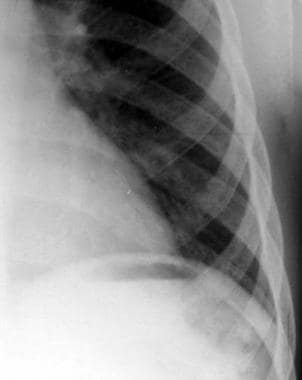 Thalassemia. Radiograph of the ribs. Note the long linear density within the medulla, which runs parallel to the long axis of the rib and is most pronounced in the mid and anterior portions of the ribs.
Thalassemia. Radiograph of the ribs. Note the long linear density within the medulla, which runs parallel to the long axis of the rib and is most pronounced in the mid and anterior portions of the ribs.
Another finding, which appears limited to the ribs, is the subcortical lucency, consisting of a sharply defined lucency at the margin of the cortex and medulla. This appears to result from erosion of the inner aspect of the cortex and adjacent medullary trabeculae by the leading edge of proliferating marrow. A highly characteristic appearance consists of bulbous expansion of the posterior and, to a lesser extent, anterior segments of the ribs, and it is frequently associated with posterior or anterior soft tissue densities, which histologically represent hematopoietic marrow.
Extramedullary hematopoiesis
In the most severely affected patients, and particularly in patients with thalassemia intermedia, prominent lobulated soft tissue densities may be noted in the posterior mediastinum and, to a lesser degree, in the anterior mediastinum or pelvis. These opacities result from extramedullary hematopoiesis (see the image below). CT scanning, which can evaluate the skeletal system in the axial plane, demonstrates that this proliferating marrow originates within the medulla of adjacent vertebral bodies, ribs, or pelvis. [6]
Thalassemia. Extramedullary hematopoiesis. Lobulated soft tissue opacities are noted overlying the ribs anteriorly and posteriorly.
Although extramedullary, it remains subperiosteal in location and, histologically, shows new bone formation. The presence of extramedullary hematopoiesis within the spinal canal may be associated with the development of spinal cord compression. [13] Rarely, new bone formation within these proliferative subperiosteal masses may be extensive enough to result in osteomatous lesions of the ribs that are apparent radiographically (see the image below). Less frequently, similar osteomata may arise from the long bones.
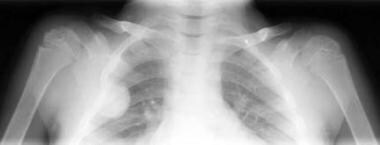 Thalassemia. Radiograph of the ribs. An osteoma is seen arising from the third rib anteriorly. The ribs are widened, are osteopenic, and demonstrate multiple areas of focal lucency.
Thalassemia. Radiograph of the ribs. An osteoma is seen arising from the third rib anteriorly. The ribs are widened, are osteopenic, and demonstrate multiple areas of focal lucency.
Vascular impressions
Hematopoietic marrow is vascular, and in thalassemia, a 5- to 30-fold increase in activity may occur as a result of marrow hyperplasia and hypertrophy. Not surprisingly, this marrow proliferation is associated with increased blood supply, which may be seen radiographically as an enlargement of the nutrient foramina of the tubular bones, particularly the phalanges.
A similar enlargement is noted in other processes associated with increased blood supply to the medullary, including sickle cell disease variants, Gaucher disease, and infectious diseases (including leprosy), as well as noninfectious inflammatory conditions such as hemophilia. The calvarial widening, which occurs in association with marrow hypertrophy, may be associated with markedly enlarged and tortuous vascular impressions of the calvarium. Rupture of associated enlarged venous structures may constitute a potential sequela of moderate cranial trauma.
Premature fusion of the epiphyses
Premature fusion of the epiphyses is a characteristic finding in thalassemia. This finding first was described by Currarino and Erlandson, [14] who noted the abnormality in 14% of 79 patients with homozygous β-thalassemia. However, all of the patients were older than 10 years, resulting in an incidence of 23% in patients older than 10 years.
The finding may be unilateral or bilateral and is noted most commonly in the proximal humerus. Less frequently, it may be identified in the distal femur, proximal tibia, and proximal femur. The fusion is associated with a variable degree of shortening and is frequently eccentric, leading to angulation of the prematurely fused epiphyses. In the proximal humerus, this is associated with a humeral varus deformity.
(See the image below.)
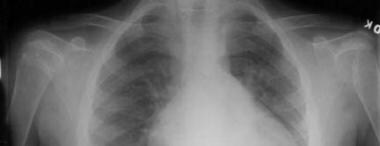 Radiograph of right and left shoulders. Fusion of the left proximal physis medially associated with a humeral varus deformity is seen. The right physis is normal.
Radiograph of right and left shoulders. Fusion of the left proximal physis medially associated with a humeral varus deformity is seen. The right physis is normal.
Currarino and Erlandson noted that a disproportionate number of patients (36%) had premature closure in the thalassemia intermedia subset of homozygous β-thalassemia. [14] An explanation for premature fusion has been suggested based on the relationship of the cortex and periosteum to the marrow proliferation (most marked in thalassemia intermedia), the fenestration of the metaphyseal cortex, the anatomy of the proximal humerus, and the forces normally exerted across the shoulder joint.
Response to therapy
The modern radiologist is unlikely to encounter radiographic thalassemic features other than in teaching files, on visits to Mediterranean countries, or in isolated patients never managed with hypertransfusion therapy. This is because therapy by hypertransfusion and chelation has improved the general health and the radiographic appearance of patients. As a radiologist, obtain a transfusional history for all patients under evaluation and be sure to understand the response of the skeletal system to therapy.
In a review of the ribs of 32 patients with homozygous β-thalassemia, the radiographic appearance of the ribs correlated with transfusional history. In this study, an overwhelming number of children hypertransfused from an early age showed normal ribs. This was in contrast to patients who were not hypertransfused until later in childhood or who were never hypertransfused, who demonstrated a spectrum of radiographic rib abnormalities, the severity of which increased with the delay in initiating transfusional therapy.
The most striking rib changes were present in patients with thalassemia intermedia who were never transfused. Regression of rib abnormalities after transfusion was not uncommon; however, in approximately two thirds of patients, regression did not occur until after the introduction of hypertransfusion therapy. The changes noted in the ribs appeared to reflect similar changes occurring in the appendicular skeleton.
Similarly, the size of the nutrient foramina in the phalanges and the vascular impressions in the calvarium are related to the age of onset and type of transfusional therapy. However, unlike the skeletal response to marrow proliferation, the enlarged vascular channels do not appear to regress with therapy but remain as a permanent record of earlier marrow changes.
Variants
In addition to the homozygous state, the thalassemic gene also may be present in the heterozygous state or in combination with some other abnormal hemoglobin (ie, HbC, HbE, HbH, HbS, HbLepore). With the exception of sickle cell thalassemia, in which radiographic changes may reflect vaso-occlusive phenomena, as well as the changes of marrow hypertrophy, the radiographic appearances of these variants are solely the result of marrow proliferation and, consequently, are dependent on the degree and duration of any anemia and the type of transfusion therapy.
The anemia that occurs in thalassemia minor, HbC thalassemia, and HbH thalassemia is usually mild; consequently, the radiographic changes typically are minimal or normal in these disorders. However, the anemia of HbE thalassemia and HbLepore thalassemia may be pronounced, with subsequent radiographic changes indistinguishable from those of homozygous β-thalassemia.
Degree of confidence
The radiographic features of thalassemia result from either a generalized marrow hypertrophy and hyperplasia, which may occur in other hemoglobinopathies, or a reticuloendothelial storage process, including Gaucher disease. These may be diagnosed radiologically with a high degree of confidence, although additional clinical and laboratory information may be required for the differential diagnosis of the hemoglobinopathies.
Magnetic Resonance Imaging
MRI measures iron indirectly by the influence of iron on nearby water molecules. Tissue iron disrupts the magnetic field for a period of time that can be measured in units of time (designated T2 or T2* by spin-echo imaging or gradient-echo imaging) or by rate (R2 and R2* by spin- and gradient-echo imaging, respectively). [1]
Cardiac T2* measurements are stratified according to risk of heart disease: >20 ms, normal; 10-20 ms, moderate or at-risk; < 10 ms, severe or high-risk. Annual T2* and echocardiography can begin at 10 years of age, given that younger children infrequently have abnormal studies. Patients with high-risk T2* should have more frequent assessments (every 6 months) until measurement shifts into a lower risk category. [5]
A meta‐analysis by Sarigianni et al found that MRI T2 and T2* are better at excluding liver iron overload than at definitively diagnosing it in individual patients. Furthermore, thresholds have not been specified and uniformly used; therefore, further studies are needed to standardize T2/T2* protocols for hepatic iron determination. [15]
Different MRI techniques may not be equivalent in the manner that they are currently calibrated and practiced. The first widely used technique was the T2* technique, where liver biopsy was used to calibrate the method. Because of factors such as long echo times and multi breath-hold acquisition, the calibration differs from later techniques and can underestimate LIC by twofold. Therefore, studies using this calibration may underestimate liver iron concentration (LIC). [7]
The R2-based Ferriscan technique has been shown to have an average sensitivity of more than 85% and a specificity of more than 92%, up to an LIC of 15 mg/g dry wt. [7]
Ultrasonography
Cholelithiasis is common in untreated thalassemia. Ultrasonography is the modality of choice when gallstones are suggested. [16]
Although ultrasonography may show gallstones in approximately 42% of sickle cell disease patients aged 15-18 years, the application of the same technique to a group of thalassemia patients maintained on hypertransfusion therapy revealed gallstones in only 3.6% of patients in a similar age group. This reduction results from the more intensive transfusional regimen, which reduces activity of the patient's bone marrow, diminishes formation of the fragile erythrocytes, and consequently limits production of excess bilirubin.
-
Thalassemia. Anteroposterior radiograph of the lumbar spine. Osteopenia is present; note striated appearance of the vertebral bodies resulting from preservation and thickening of the vertical trabeculae. Clips from a previous cholecystectomy are present.
-
Thalassemia. Lateral skull radiograph. Widening of the calvarium is present; the outer table overlying the frontal region is obliterated and new bone formation can be seen in the diploë, producing a "hair-on-end" appearance. Note that the occipital bone is uninvolved. The impressions of the calvarial vessels are enlarged.
-
Thalassemia. Anteroposterior of the hands. A generalized loss of bone density is observed. The cortex is thinned, and the trabeculae are coarsened and outline localized lucency. Widening of the medullary cavity has resulted in squaring of the metacarpals. Note the enlarged nutrient foramina in the middle phalanges.
-
Thalassemia. Anteroposterior radiograph of the forearm. A fracture is noted in the distal radius. Evidence of medullary expansion and cortical thinning is observed.
-
Thalassemia. Radiograph of the ribs. Erosions of the inferior cortical margins of the third, fourth, and fifth ribs are noted.
-
Thalassemia. Radiograph of the ribs. Note the long linear density within the medulla, which runs parallel to the long axis of the rib and is most pronounced in the mid and anterior portions of the ribs.
-
Thalassemia. Extramedullary hematopoiesis. Lobulated soft tissue opacities are noted overlying the ribs anteriorly and posteriorly.
-
Thalassemia. Radiograph of the ribs. An osteoma is seen arising from the third rib anteriorly. The ribs are widened, are osteopenic, and demonstrate multiple areas of focal lucency.
-
Radiograph of right and left shoulders. Fusion of the left proximal physis medially associated with a humeral varus deformity is seen. The right physis is normal.








