Practice Essentials
Autosomal dominant polycystic kidney disease (ADPKD) is a hereditary disorder. Because the disorder occurs equally in males and females, each offspring has a 50% chance of inheriting the responsible mutation and therefore the disease. ADPKD is a genetically heterogeneous condition that involves at least 2 genes: PKD1 accounts for most ADPKD cases, and PKD2 iaccounts for 15% of cases. Cysts arise from the nephrons and collecting tubules; microdissection reveals that the cysts communicate directly with the nephrons and collecting tubules. Islands of normal parenchymal renal tissue are interspaced between the cysts. [1, 2, 3, 4, 5]
Patients with ADPKD present with hypertension and progressive renal failure after their third decade of life. The disease is uncommon in children and is rarely seen in neonates. Gardner and Evan showed that individuals older than age 40 years with a family history of ADPKD but without renal cysts are unlikely to develop the disease. Other researchers have shown that if the kidney shows no signs of cysts or parenchymal abnormality in a patient by age 19 years, that individual is extremely unlikely to be affected. [1, 2, 3, 4, 5]
ADPKD has historically been estimated to affect 1 in 1000 people; however, an analysis of population whole-genome sequencing suggests that the prevalence of ADPKD may be higher. [6] Between 29% and 73% of patients with the disorder have associated hepatic cysts, 9% have associated pancreatic cysts, and 5% have associated splenic cysts; pulmonary cysts occur but are uncommon. Intracranial aneurysms affect 10-11.5% of patients with ADPKD, which is approximately 5 times higher than the rate in the general population. When aneurysms erupt, the morbidity and mortality rates range from 35-55%. [7] MR angiography screening of patients with ADPKD every 5 years and annual follow-up in patients with detected intracranial aneurysm has been reported to be a cost-effective management strategy. [8, 9, 10, 11, 12, 13, 14, 15, 16]
(For examples of polycystic kidney disease, see the images below.)
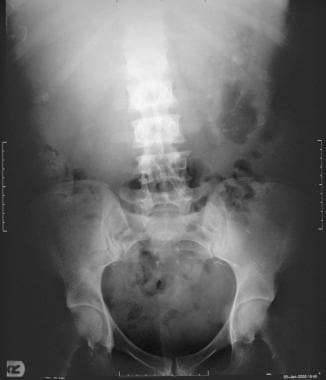 Plain radiograph of the kidney, ureters, and bladder in a 50-year-old woman with autosomal dominant polycystic kidney disease. The kidneys are enlarged with multiple curvilinear and ringlike calcifications arising from the renal cyst. The surgical clip from renal transplant is seen projected over the left iliac wing.
Plain radiograph of the kidney, ureters, and bladder in a 50-year-old woman with autosomal dominant polycystic kidney disease. The kidneys are enlarged with multiple curvilinear and ringlike calcifications arising from the renal cyst. The surgical clip from renal transplant is seen projected over the left iliac wing.
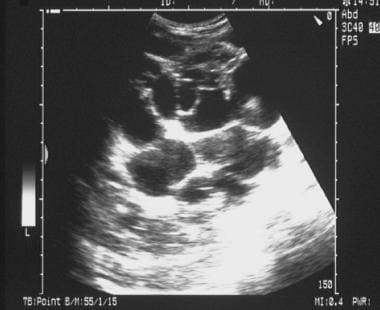 Sonogram of the kidney in a patient with polycystic kidney disease shows numerous cysts of varying sizes.
Sonogram of the kidney in a patient with polycystic kidney disease shows numerous cysts of varying sizes.
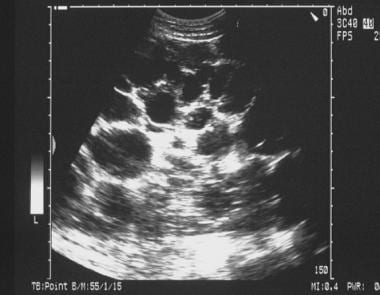 Sonogram of the liver in a patient with polycystic kidney disease shows multiple cysts. Approximately 29-73% of patients with autosomal dominant polycystic kidney disease have cysts in the liver.
Sonogram of the liver in a patient with polycystic kidney disease shows multiple cysts. Approximately 29-73% of patients with autosomal dominant polycystic kidney disease have cysts in the liver.
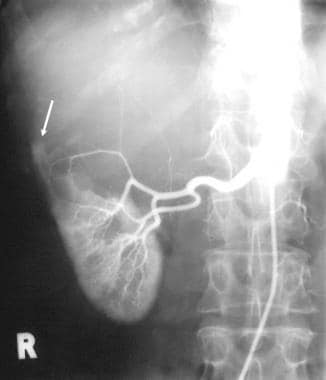
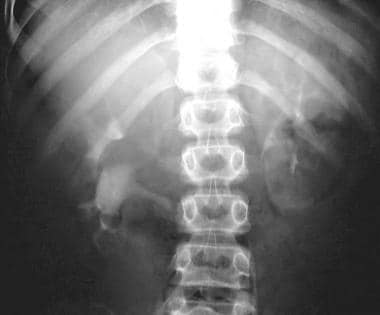 Excretory 5-minute urographic image in a young male patient with bilateral polycystic disease. The calyces are elongated and splayed because of the cysts, seen best on the right. Note the large size of both kidneys.
Excretory 5-minute urographic image in a young male patient with bilateral polycystic disease. The calyces are elongated and splayed because of the cysts, seen best on the right. Note the large size of both kidneys.
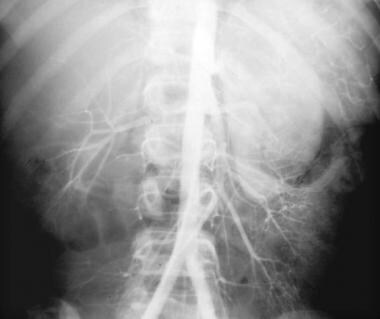 Aortogram in a young male patient with bilateral polycystic disease demonstrates stretching of the intrarenal arterial branches, seen best in the upper pole of the right kidney.
Aortogram in a young male patient with bilateral polycystic disease demonstrates stretching of the intrarenal arterial branches, seen best in the upper pole of the right kidney.
Radiologic examination
Ultrasonography is the procedure of choice in the workup of patients with ADPKD, and it is an ideal modality for screening patients' families (see the images below). Plain radiographs offer limited information. Plain radiographic findings are normal in the early stages of ADPKD, but with enlargement of the kidneys, soft-tissue masses displace the intra-abdominal organs.
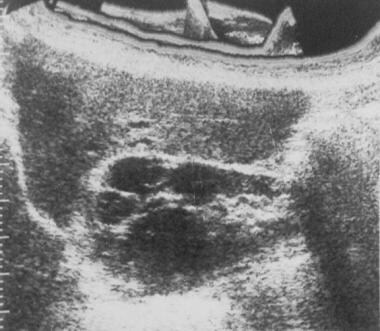 Sonogram of the right kidney in a patient with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of varying sizes, with a large cyst in the upper pole.
Sonogram of the right kidney in a patient with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of varying sizes, with a large cyst in the upper pole.
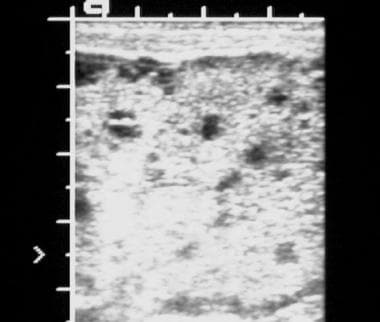 Sonogram of the liver in a newborn with polycystic kidney disease shows numerous tiny cysts affecting both lobes of the liver.
Sonogram of the liver in a newborn with polycystic kidney disease shows numerous tiny cysts affecting both lobes of the liver.
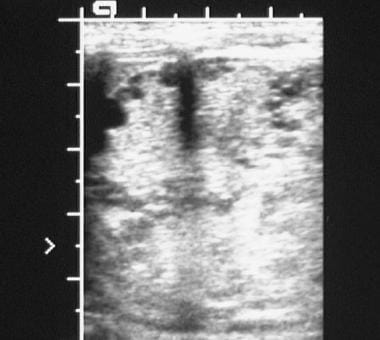 Sonogram of the kidney in a newborn with polycystic kidney disease shows numerous cysts of varying sizes, predominantly situated in the periphery.
Sonogram of the kidney in a newborn with polycystic kidney disease shows numerous cysts of varying sizes, predominantly situated in the periphery.
In studies in young children, intravenous urography and nephrotomography were shown to be slightly more sensitive than ultrasonography.
Computed tomography (CT) scanning is as sensitive as ultrasonography in the detection of cystic disease, although problems may arise with smaller cysts. CT scanning appears to be more specific than ultrasonography in differentiating an obstructed renal pelvis from a parapelvic cyst. It also seems to be superior to ultrasonographic images in helping to assess retroperitoneal rupture of a cyst and perinephric extension of blood or pus from an infected cyst. [17, 18] [19]
Magnetic resonance imaging (MRI) is especially useful for examining patients who are allergic to iodinated contrast media and those with compromised renal function who are at risk for iodinated contrast–induced renal failure. MRI also has advantages for patients in whom hemorrhagic cysts are considered and is probably superior to other modalities in characterizing complicated cysts. [20, 21, 22, 23]
Radionuclide studies have a complementary role in the assessment of renal function in ADPKD. These studies do not have the added hazard of patient exposure to iodinated contrast material.
Nephromegaly, which can be detected on plain radiographs, may result from causes other than ADPKD. Similarly, curvilinear calcification is not specific for ADPKD and may be found in other types of cysts, as well as in tumors and granulomas. Simple renal cysts with nongenetic origins may be similar to ADPKD lesions. Also, cysts associated with ADPKD cannot always be differentiated from multiple simple cysts and cysts associated with von Hippel–Lindau disease or tuberous sclerosis. Such findings apply to intravenous urography (seen in the image below), ultrasonography, CT scanning, MRI, and angiography.
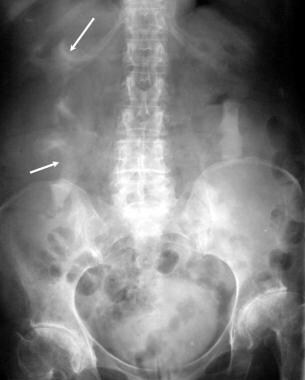 Excretory 30-minute urographic image in a patient with polycystic kidney disease. The kidneys are enlarged with an elongated, splayed, and distorted collecting system resulting from the presence of innumerable cysts. Note bilateral avascular necrosis of the femoral heads.
Excretory 30-minute urographic image in a patient with polycystic kidney disease. The kidneys are enlarged with an elongated, splayed, and distorted collecting system resulting from the presence of innumerable cysts. Note bilateral avascular necrosis of the femoral heads.
Imaging Guidelines
An international working group has issued a consensus statement on imaging of ADPKD in children. The consensus statement recommended that in patients younger than aged 15 years with a positive family history, the presence of at least one kidney cyst, kidney enlargement above normal limits, or both should be considered highly suggestive of ADPKD. Additional recommendations included the following [24] :
-
Ultrasonography (US) is the most useful imaging modality with which to diagnose ADPKD.
-
Although high-resolution US and MRI are able to depict more and smaller cysts than can standard US in adults, there are no diagnostic criteria for their use in children.
-
In children with a negative family history or very-early onset ADPKD, the diagnosis should be confirmed with genetic testing.
-
Progression of ADPKD should be monitored in cooperative children with MRI to determine total kidney volume; US monitoring of kidney size and cyst number is preferable to MRI in uncooperative children.
Radiography
Plain radiographic findings are normal in the early stages of autosomal dominant polycystic kidney disease (ADPKD); however, with enlargement of the kidneys, soft-tissue masses that displace intra-abdominal organs may be seen.
Renal enlargement is often asymmetrical, and usually, normal renal outlines cannot be traced.
Cysts may calcify in a curvilinear, ringlike, or amorphous manner (see the image below). The presence of renal calculi may signify urinary tract infection.
Plain radiograph of the kidney, ureters, and bladder in a 50-year-old woman with autosomal dominant polycystic kidney disease. The kidneys are enlarged with multiple curvilinear and ringlike calcifications arising from the renal cyst. The surgical clip from renal transplant is seen projected over the left iliac wing.
In younger, asymptomatic patients, the kidneys may be smoothly enlarged, with normal collecting systems depicted by intravenous urography; eventually, however, the renal contour becomes lobulated.
Nephrogram findings may have a characteristic Swiss cheese appearance resulting from numerous, smoothly marginated radiolucencies distributed throughout the renal cortex and medulla.
The pelvocaliceal systems demonstrate bilateral, diffuse irregularity and splaying. The collecting system may be elongated, distorted, and attenuated (see the image below).
Excretory 30-minute urographic image in a patient with polycystic kidney disease. The kidneys are enlarged with an elongated, splayed, and distorted collecting system resulting from the presence of innumerable cysts. Note bilateral avascular necrosis of the femoral heads.
Large cysts may occasionally obstruct a calyx.
Occasionally, cysts may rupture into the collecting system and appear, on intravenous urography or retrograde pyelography, as a contrast material–filled cavity communicating with a calyx.
In ADPKD, the kidneys shrink with the onset of renal failure, after renal transplantation, or as a result of long-term hemodialysis.
Uncommonly, patients with well-developed renal cysts, even patients with chronic renal failure, have normal-sized kidneys.
Delayed, diffuse, nodular puddling of contrast material throughout the renal parenchyma has been reported in neonates with ADPKD. This phenomenon is said to be useful in differentiating ADPKD from autosomal recessive polycystic kidney disease (ARPKD). In ARPKD, the nephrogram usually shows striation, with persistent radiating opaque streaks on delayed images (sunburst nephrogram).
Retrograde pyelography may demonstrate normal findings when the cysts are small. With larger cysts, this modality may reveal splaying, effacement, and, uncommonly, dilatation of the calyces and the renal pelvis. Some cysts may fill with contrast material.
Degree of confidence
Plain abdominal radiographs have low specificity and generally are not recommended in the workup of patients with ADPKD.
Differentiating between multiple simple renal cysts and the cysts of ADPKD may be difficult with intravenous urography.
Detectable on plain radiographs, nephromegaly may result from causes other than ADPKD. Similarly, curvilinear calcification may occur in simple renal cysts and tumors. Simple renal cysts with nongenetic origins can resemble ADPKD cysts, as can lesions arising from von Hippel–Lindau disease or tuberous sclerosis.
Computed Tomography
On CT scans, the cysts in autosomal dominant polycystic kidney disease (ADPKD) are fairly well-defined, round or oval masses with low attenuation values similar to those of water (see the images below).
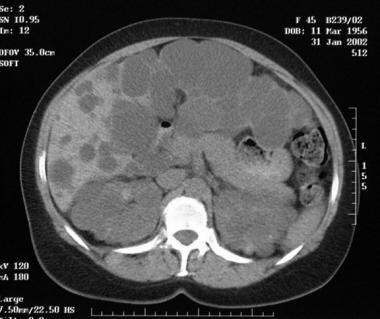 Unenhanced axial computed tomography scan of the abdomen in a 45-year-old woman with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of different sizes involving the kidneys, liver, and pancreas.
Unenhanced axial computed tomography scan of the abdomen in a 45-year-old woman with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of different sizes involving the kidneys, liver, and pancreas.
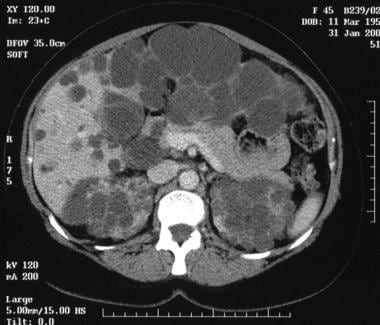 Contrast-enhanced computed tomography scan in a 45-year-old woman with autosomal dominant polycystic kidney disease (same patient as in the previous image) clearly demonstrates the cysts in the head of the pancreas.
Contrast-enhanced computed tomography scan in a 45-year-old woman with autosomal dominant polycystic kidney disease (same patient as in the previous image) clearly demonstrates the cysts in the head of the pancreas.
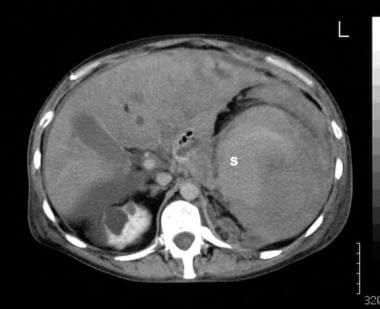 A 42-year-old man known to have autosomal dominant polycystic kidney disease presented with sudden left-upper-quadrant pain and hypotension. Sonography performed in the emergency department showed echogenic fluid in the left upper quadrant. The spleen was not identified. Some free peritoneal fluid also was seen (sonogram not shown). Contrast-enhanced computed tomography scan of the upper abdomen shows that the medial spleen (S) is associated with both intracapsular and extracapsular fluid. Note the cysts within the right kidney.
A 42-year-old man known to have autosomal dominant polycystic kidney disease presented with sudden left-upper-quadrant pain and hypotension. Sonography performed in the emergency department showed echogenic fluid in the left upper quadrant. The spleen was not identified. Some free peritoneal fluid also was seen (sonogram not shown). Contrast-enhanced computed tomography scan of the upper abdomen shows that the medial spleen (S) is associated with both intracapsular and extracapsular fluid. Note the cysts within the right kidney.
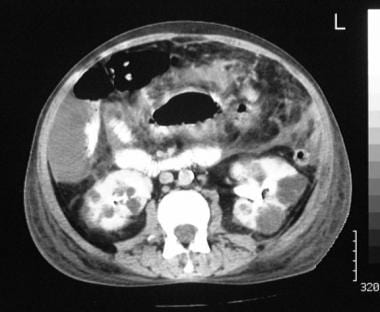 Contrast-enhanced computed tomography scan of the upper abdomen in the same patient as in the previous image shows cysts in both kidneys. Note the free peritoneal fluid around the tip of the liver.
Contrast-enhanced computed tomography scan of the upper abdomen in the same patient as in the previous image shows cysts in both kidneys. Note the free peritoneal fluid around the tip of the liver.
After the intravenous administration of iodinated contrast material, cysts do not enhance but instead stand out prominently against normally enhancing background renal tissue.
Obstruction of a calyx or, rarely, the renal pelvis may occur. In the early disease stages, renal size may be normal and the reniform shape of the kidneys is well maintained.
With advancing disease, the size and number of cysts increase and the renal outline may become scalloped, with the cyst projecting beyond the renal outline.
With end-stage renal disease, unenhanced CT scans may depict curvilinear or amorphous calcification within the walls of the cyst. Renal calculi are well depicted on CT scans.
Acute hemorrhage into a cyst is shown as a high-attenuation mass on unenhanced scans in patients presenting with acute flank pain. These hemorrhagic cysts are usually subcapsular. With time, the hemorrhage resolves and the attenuation of the cystic contents is reduced.
Spontaneous hemorrhage into the retroperitoneum is a known complication that is demonstrated readily on CT scans.
Infected cysts usually have thick, irregular walls that often calcify. The attenuation value of an infected cyst is slightly higher than that of water; the value may vary within the cyst because of the presence of debris.
Localized thickening of the Gerota fascia may be associated with cyst infection and renal abscess.
Degree of confidence
CT scanning is a sensitive modality for examining cystic kidney disease. However, problems may arise with smaller cysts, which may not be easily differentiated from small, solid masses. Occasionally, differentiation of chronically infected cysts from a necrotic tumor also may be difficult.
CT scanning appears to be more specific than ultrasonography in differentiating an obstructed renal pelvis from a parapelvic cyst on the basis of contrast excretion pattern. It is also useful in helping assess retroperitoneal rupture of a cyst and perinephric extension of blood or pus from an infected cyst.
As with images derived from intravenous urography, CT scans of lesions associated with ADPKD can resemble CT scans of simple renal cysts of nongenetic etiology, as well as CT scans of cysts associated with either von Hippel–Lindau disease or tuberous sclerosis.
Magnetic Resonance Imaging
Renal cysts show a homogeneous, low to intermediate signal intensity on T1-weighted images and a homogeneous, high signal intensity on T2-weighted images.
Cysts have thin, imperceptible walls. Renal cysts do not enhance with gadolinium-based contrast material.
Gadolinium-based contrast agents have been linked to the development of nephrogenic systemic fibrosis (NSF) or nephrogenic fibrosing dermopathy (NFD). The disease has occurred in patients with moderate to end-stage renal disease after being given a gadolinium-based contrast agent to enhance MRI or magnetic resonance angiography (MRA) scans. NSF/NFD is a debilitating and sometimes fatal disease. Characteristics include red or dark patches on the skin; burning, itching, swelling, hardening, and tightening of the skin; yellow spots on the whites of the eyes; joint stiffness with trouble moving or straightening the arms, hands, legs, or feet; pain deep in the hip bones or ribs; and muscle weakness.
As they do when imaged in other modalities, kidneys affected by ADPKD appear large on MRI scans and have irregular, distorted contours.
Uncomplicated cysts resemble simple cysts.
MRI can be used to help characterize renal cysts that are equivocal with other imaging modalities; such cysts include hyperintense cysts, hemorrhagic cysts, and cysts with calcification or septa.
Cysts complicated by hemorrhage may look hyperintense on all image sequences, but appearances may depend on the age of the hemorrhage.
Layering may be present with a recent intracystic bleed.
Cysts complicated by infection or those with high protein content may present with an intermediate signal intensity on T1-weighted or T2-weighted images. [21, 22, 23]
Degree of confidence
MRI of the kidneys is a useful technique for characterization of renal masses and can be used in addition to or instead of CT scanning. MRI is especially useful in patients who are allergic to iodinated contrast media and in those with compromised renal function who are at risk for iodinated contrast–induced renal failure. MRI has the added advantage of multiplanar imaging and does not require the use of ionizing radiation. Moreover, MRI is probably superior to other modalities in characterizing complicated cysts. [25]
As with images derived from intravenous urography and CT scanning, MRI scans of simple renal cysts of nongenetic etiology, as well as those of cysts associated with either von Hippel–Lindau disease or tuberous sclerosis, can resemble scans of lesions associated with ADPKD.
Ultrasonography
Ultrasonography is the procedure of choice in the workup of patients with ADPKD. It is also an ideal modality for screening the family of patients with known disease and for routine follow-up in patients. Cysts that are a few millimeters in size can be depicted. [26, 27]
Sequential examinations usually demonstrate an increase in cyst size and number over a period of years, resulting in calyceal distortion and irregularity of the renal outline.
Cysts may have irregular walls and may show internal echoes if complications of hemorrhage or infection are present.
Surviving islands of renal parenchyma may be seen between cysts. Calcification of cyst walls may be seen later in life.
Renal cysts similar to those associated with ADPKD can occur as simple renal cysts without genetic etiology or can arise in von Hippel–Lindau disease, in acquired uremic cystic disease (in which kidneys are small, with no renal function), in patients on hemodialysis, or in patients who have had renal transplantation.
In ARPKD, the kidneys are hyperechoic and enlarged, but the cysts are too small to be depicted on ultrasonographic images; however, discrete macroscopic cysts (usually < 1 cm) occasionally are seen in patients with ARPKD.
Most children with autosomal dominant polycystic kidney disease (ADPKD) have normal ultrasonographic findings at birth.
In neonates who are severely affected, the kidneys are large and echogenic, with loss of corticomedullary differentiation indistinguishable from that of ARPKD.
(See the image below.)
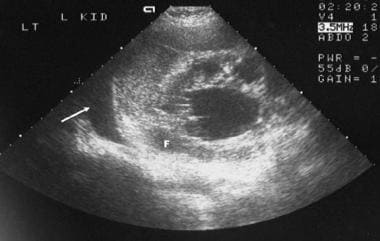 Sonogram of the left kidney in a 45-year-old woman with bilateral polycystic kidney disease who presented with acute onset of left loin pain. The scan shows fluid around the left kidney and in the left pleural space, consistent with a ruptured renal cyst.
Sonogram of the left kidney in a 45-year-old woman with bilateral polycystic kidney disease who presented with acute onset of left loin pain. The scan shows fluid around the left kidney and in the left pleural space, consistent with a ruptured renal cyst.
When cysts are seen, they vary in size (as seen in the images below) and are scattered throughout the cortex, including in medullar and subcapsular locations. By contrast, the peripheral cortex is spared in patients with mild ARPKD.
Sonogram of the kidney in a patient with polycystic kidney disease shows numerous cysts of varying sizes.
Sonogram of the right kidney in a patient with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of varying sizes, with a large cyst in the upper pole.
Sonogram of the kidney in a newborn with polycystic kidney disease shows numerous cysts of varying sizes, predominantly situated in the periphery.
Cysts of the liver, pancreas, and spleen may help to confirm the diagnosis of ADPKD (see the images below).
Sonogram of the liver in a patient with polycystic kidney disease shows multiple cysts. Approximately 29-73% of patients with autosomal dominant polycystic kidney disease have cysts in the liver.
Sonogram of the liver in a newborn with polycystic kidney disease shows numerous tiny cysts affecting both lobes of the liver.
ADPKD is a rare occurrence in the fetus. The kidneys may be enlarged as a result of cystic dilatation of tubules and glomeruli, and this is associated with increased echogenicity mimicking autosomal recessive disease. Occasionally, the cortical cysts are large enough to be detected on ultrasonographic images; this can confirm the diagnosis when seen in the fetus of a parent with the disease. Since renal function in utero is adequate, oligohydramnios is not present.
Nuclear Imaging
Technetium-99m (99mTc) mercaptotriglycylglycine or other radionuclides are employed in isotope renography, which is a useful procedure for assessing renal function and avoiding the nephrotoxic effects of iodinated contrast media. [28] On analog images, cysts are demonstrated as photon-deficient masses.
Similarly, uptake of 99mTc dimercaptosuccinic acid shows the cysts as multiple photon-deficient masses.
Hepatic and splenic cysts also are photopenic on scans using 99mTc sulfur colloid. Infected cysts, when scanned with granulocytes labeled with 99mTc hexamethylpropyleneamine oxime, may demonstrate increased activity. Similarly, they may be avid for gallium-67 citrate. [20]
Although isotopic renography is an excellent modality for assessing absolute and relative renal function, its role in the diagnosis of autosomal dominant polycystic kidney disease is limited.
Multiple photon-deficient renal masses may occur with simple cysts, granulomas, abscesses, and multiple tumors.
Angiography
Angiography has a high degree of sensitivity in the diagnosis of autosomal dominant polycystic kidney disease (ADPKD), but its specificity is low.
Differentiation from simple renal cysts is not always possible; avascular tumors also may pose problems. Renal sinus fibrolipomatosis may superficially resemble cysts associated with ADPKD.
Angiographic criteria for cystic renal disease are well established. In the arterial phase, intrarenal arterial branches are stretched and displaced around the cysts, as seen in the image below.
Aortogram in a young male patient with bilateral polycystic disease demonstrates stretching of the intrarenal arterial branches, seen best in the upper pole of the right kidney.
The cysts are seen as sharply defined, radiolucent masses. The cyst walls appear thin and smooth, and an acute angle is characteristic between the cyst and the normal renal cortex, forming a beak (as seen in the image below). In the nephrogram phase, avascular areas of varying sizes are demonstrated, giving rise to a Swiss cheese appearance, as on an intravenous urogram.
 Selective renal arteriogram in a patient with autosomal dominant polycystic kidney disease. A large filling defect is demonstrated in the upper pole of the right kidney, forming an acute angle with the normal renal cortex that results in a characteristic beak appearance.
Selective renal arteriogram in a patient with autosomal dominant polycystic kidney disease. A large filling defect is demonstrated in the upper pole of the right kidney, forming an acute angle with the normal renal cortex that results in a characteristic beak appearance.
Hemorrhagic and infected cysts may have abnormal vessels simulating the neovascularity of a neoplasm. Hepatic and splenic cysts appear as avascular masses on a flush aortogram or on selective visceral angiography (see the image below).
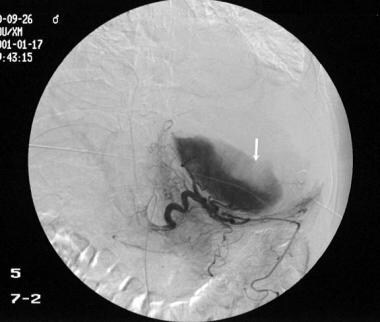 Because this 42-year-old man was known to have autosomal dominant polycystic kidney disease and was hemodynamically unstable, a splenic angiogram was obtained, with a focus on embolizing the spleen. The angiogram shows a medially displaced spleen with separation or stretching of the splenic capsule. A slit is seen in the lower lateral border as a result of splenic rupture (arrow). The spleen was embolized successfully, and no surgical intervention was required. The patient had splenic cysts, as previously recorded elsewhere. The presumptive diagnosis was spontaneous rupture of a splenic cyst. The patient had no history of trauma.
Because this 42-year-old man was known to have autosomal dominant polycystic kidney disease and was hemodynamically unstable, a splenic angiogram was obtained, with a focus on embolizing the spleen. The angiogram shows a medially displaced spleen with separation or stretching of the splenic capsule. A slit is seen in the lower lateral border as a result of splenic rupture (arrow). The spleen was embolized successfully, and no surgical intervention was required. The patient had splenic cysts, as previously recorded elsewhere. The presumptive diagnosis was spontaneous rupture of a splenic cyst. The patient had no history of trauma.
-
Plain radiograph of the kidney, ureters, and bladder in a 50-year-old woman with autosomal dominant polycystic kidney disease. The kidneys are enlarged with multiple curvilinear and ringlike calcifications arising from the renal cyst. The surgical clip from renal transplant is seen projected over the left iliac wing.
-
Excretory 30-minute urographic image in a patient with polycystic kidney disease. The kidneys are enlarged with an elongated, splayed, and distorted collecting system resulting from the presence of innumerable cysts. Note bilateral avascular necrosis of the femoral heads.
-
Sonogram of the kidney in a patient with polycystic kidney disease shows numerous cysts of varying sizes.
-
Sonogram of the liver in a patient with polycystic kidney disease shows multiple cysts. Approximately 29-73% of patients with autosomal dominant polycystic kidney disease have cysts in the liver.
-
Excretory 5-minute urographic image in a young male patient with bilateral polycystic disease. The calyces are elongated and splayed because of the cysts, seen best on the right. Note the large size of both kidneys.
-
Aortogram in a young male patient with bilateral polycystic disease demonstrates stretching of the intrarenal arterial branches, seen best in the upper pole of the right kidney.
-
Sonogram of the right kidney in a patient with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of varying sizes, with a large cyst in the upper pole.
-
Selective renal arteriogram in a patient with autosomal dominant polycystic kidney disease. A large filling defect is demonstrated in the upper pole of the right kidney, forming an acute angle with the normal renal cortex that results in a characteristic beak appearance.
-
Sonogram of the liver in a newborn with polycystic kidney disease shows numerous tiny cysts affecting both lobes of the liver.
-
Sonogram of the kidney in a newborn with polycystic kidney disease shows numerous cysts of varying sizes, predominantly situated in the periphery.
-
Unenhanced axial computed tomography scan of the abdomen in a 45-year-old woman with autosomal dominant polycystic kidney disease. The scan shows numerous cysts of different sizes involving the kidneys, liver, and pancreas.
-
Contrast-enhanced computed tomography scan in a 45-year-old woman with autosomal dominant polycystic kidney disease (same patient as in the previous image) clearly demonstrates the cysts in the head of the pancreas.
-
Sonogram of the left kidney in a 45-year-old woman with bilateral polycystic kidney disease who presented with acute onset of left loin pain. The scan shows fluid around the left kidney and in the left pleural space, consistent with a ruptured renal cyst.
-
A 42-year-old man known to have autosomal dominant polycystic kidney disease presented with sudden left-upper-quadrant pain and hypotension. Sonography performed in the emergency department showed echogenic fluid in the left upper quadrant. The spleen was not identified. Some free peritoneal fluid also was seen (sonogram not shown). Contrast-enhanced computed tomography scan of the upper abdomen shows that the medial spleen (S) is associated with both intracapsular and extracapsular fluid. Note the cysts within the right kidney.
-
Contrast-enhanced computed tomography scan of the upper abdomen in the same patient as in the previous image shows cysts in both kidneys. Note the free peritoneal fluid around the tip of the liver.
-
Because this 42-year-old man was known to have autosomal dominant polycystic kidney disease and was hemodynamically unstable, a splenic angiogram was obtained, with a focus on embolizing the spleen. The angiogram shows a medially displaced spleen with separation or stretching of the splenic capsule. A slit is seen in the lower lateral border as a result of splenic rupture (arrow). The spleen was embolized successfully, and no surgical intervention was required. The patient had splenic cysts, as previously recorded elsewhere. The presumptive diagnosis was spontaneous rupture of a splenic cyst. The patient had no history of trauma.






