Practice Essentials
Sickle cell disease (SCD) is a chronic hemoglobinopathy of clinical relevance because of its significant morbidity and mortality, particularly in people of African and Mediterranean ancestry. SCD results from a mutated form of hemoglobin, hemoglobin S (HBS), and vascular obstruction and ischemia result in a wide range of clinical problems. [1, 2, 3, 4, 5] Effects on the musculoskeletal system include extramedullary hematopoiesis, osteonecrosis, dactylitis (hand-foot syndrome), myonecrosis, and osteomyelitis,with bone disease being one of the most common presentations in patients with SCD. [6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16]
Preferred examination
MRI is the best method for detecting early signs of changes in bone marrow due to acute and chronic bone marrow infarction, marrow hyperplasia, osteomyelitis, and osteonecrosis. [5, 17, 18, 19, 20, 11] Differentiation of bone infarct from osteomyelitis is one of the most challenging issues in the evaluation of acute bone pain in patients with SCD. Atypical findings on diffusion-weighted images have been identified in the early phase of presentation and can help differentiate bone infarct from osteomyelitis. [11]
(See the SCD images below.)
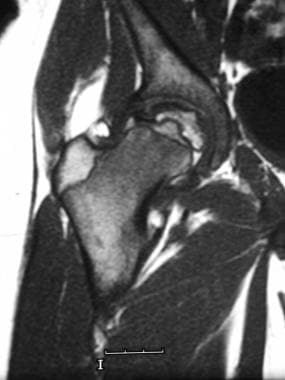 Skeletal sickle cell anemia. Osteonecrosis. Coronal T1-weighted MRI shows a slightly flattened femoral head with a serpentine margin of low signal intensity around an area of ischemic marrow with signal intensity similar to that of fat.
Skeletal sickle cell anemia. Osteonecrosis. Coronal T1-weighted MRI shows a slightly flattened femoral head with a serpentine margin of low signal intensity around an area of ischemic marrow with signal intensity similar to that of fat.
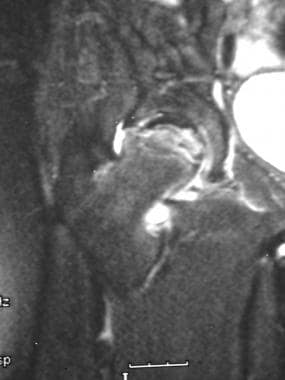 Skeletal sickle cell anemia. Osteonecrosis in the same patient as in the previous image. Coronal T2-weighted MRI shows a serpentine area of low signal intensity and additional focal areas of abnormal low signal intensity in the femoral head; these findings reflect collapse of bone and sclerosis.
Skeletal sickle cell anemia. Osteonecrosis in the same patient as in the previous image. Coronal T2-weighted MRI shows a serpentine area of low signal intensity and additional focal areas of abnormal low signal intensity in the femoral head; these findings reflect collapse of bone and sclerosis.
Nuclear imaging can also be used to detect early osteonecrosis (see the image below). This modality also plays a role in detecting osteomyelitis. Likewise, indium leukocyte scanning has an important role in diagnosing osteomyelitis.
 Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.
Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.
Plain radiography of the extremities is useful in evaluating subacute and chronic infarction and in assessing the number and severity of prior episodes of infarction (see the images below). Plain radiographs are also excellent for evaluating deformities and other complications of bone infarction.
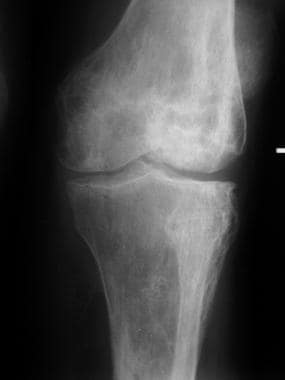 Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
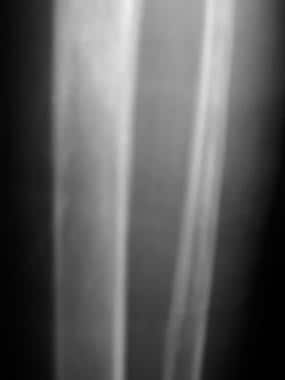 Skeletal sickle cell anemia. Medullary sclerosis. Image shows patchy sclerosis of the proximal tibia due to old infarctions. In other cases, sclerosis may be diffuse rather than patchy.
Skeletal sickle cell anemia. Medullary sclerosis. Image shows patchy sclerosis of the proximal tibia due to old infarctions. In other cases, sclerosis may be diffuse rather than patchy.
Osteonecrosis is visible on plain images only in the later stages after the affected bone is substantially damaged.
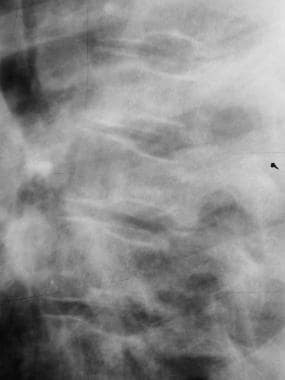 Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
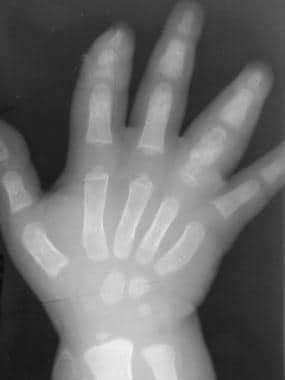 Skeletal sickle cell anemia. Hand-foot syndrome. Soft tissue swelling with periosteal new-bone formation and a moth-eaten lytic process at the proximal aspect of the fourth phalanx.
Skeletal sickle cell anemia. Hand-foot syndrome. Soft tissue swelling with periosteal new-bone formation and a moth-eaten lytic process at the proximal aspect of the fourth phalanx.
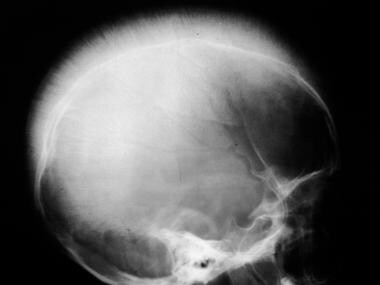 Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
Ever since carriers of the mutated gene survived the deadly malaria epidemics that are thought to have occurred thousands of years ago, the gene has continued to survive in malaria-endemic areas. However, in areas such as the United States, where malaria is not a problem, the trait no longer provides a survival advantage and instead poses the threat of sickle cell disease if the carrier's children inherit the sickle cell gene from both parents (ie, HbSS). Although carriers of sickle cell trait do not suffer from the disease, individuals with one copy of HbS and one copy of a different beta-globin gene variant such as HbC or Hb beta-thalassemia have a less severe form of the disease.
Although the disease is most frequently found in sub-Saharan Africa, it is also found in some parts of Sicily, Greece, southern Turkey, and India, all of which have areas in which malaria is endemic.
Manifestations of sickle cell disease
The skeletal manifestations of sickle cell disease are the result of changes in bone and bone marrow caused by the chronic tissue hypoxia that is exacerbated by episodic occlusion of the microcirculation by the abnormal sickle cells. The main processes that lead to bone and joint destruction in sickle cell disease are infarction of bone and bone marrow, compensatory bone marrow hyperplasia, secondary osteomyelitis, and secondary growth defects. [6, 7, 8, 9, 10, 19, 11, 12, 13, 14, 15, 16]
When the rigid erythrocytes jam in the arterial and venous sinusoids of skeletal tissue, the resultant effect is intravascular thrombosis, which leads to infarction of bone and bone marrow. Repeated episodes of these crises eventually lead to irreversible bone infarcts and osteonecrosis, especially in weight-bearing areas. These areas of osteonecrosis (avascular necrosis/aseptic necrosis) become radiographically visible as sclerosis of bone with secondary reparative reaction and eventually result in degenerative bone and joint destruction.
Infarction of bone and bone marrow in patients with sickle cell disease can lead to the following changes: osteolysis (in acute infarction), osteonecrosis (avascular necrosis/aseptic necrosis), articular disintegration, myelosclerosis, periosteal reaction (unusual in the adult), H vertebrae (steplike endplate depression also known as the Reynold sign or codfish vertebrae) (see the first image below), dystrophic medullary calcification, and bone-within-bone appearance.
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
 Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
The shortened survival time of the erythrocytes in sickle cell (10-20 days) leads to a compensatory marrow hyperplasia throughout the skeleton. The bone marrow hyperplasia has the resultant effect of weakening the skeletal tissue by widening the medullary cavities, replacing trabecular bone and thinning cortices.
Deossification due to marrow hyperplasia can bring about the following changes in bone: decreased density of skull, decreased thickness of outer table of skull due to widening of diploe, hair on-end striations of the calvaria (see the images below), and osteoporosis sometimes leading to biconcave vertebrae, coarsening of trabeculae in long and flat bones, and pathologic fractures. [20, 21]
Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
 Skeletal sickle cell anemia. Detailed view of the expanded medullary cavity in the same patient as in the previous image.
Skeletal sickle cell anemia. Detailed view of the expanded medullary cavity in the same patient as in the previous image.
Patients with sickle cell disease can have a variety of growth defects due to the abnormal maturation of bone. The following growth defects are often seen in sickle cell disease: bone shortening (premature epiphyseal fusion) (see the image below), epiphyseal deformity with cupped metaphysis, peg-in-hole defect of distal femur, and decreased height of vertebrae (short stature and kyphoscoliosis).
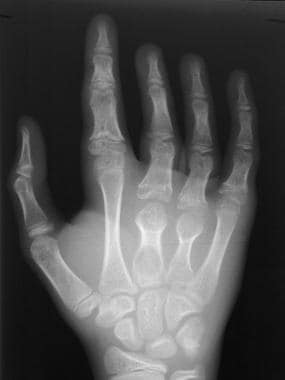 Skeletal sickle cell anemia. Bone deformity. Image shows shortening of the second and third metacarpals and phalanges with partial or complete early fusion of the growth plates due to osteonecrosis in infancy. Osteomyelitis is now superimposed the third metacarpal.
Skeletal sickle cell anemia. Bone deformity. Image shows shortening of the second and third metacarpals and phalanges with partial or complete early fusion of the growth plates due to osteonecrosis in infancy. Osteomyelitis is now superimposed the third metacarpal.
Interventional radiology
Interventional radiologists may play a role in obtaining a sample to identify the infecting organism in osteomyelitis. Also, patients with subperiosteal and soft tissue abscesses may benefit from imaging guided drainage with light sedation, avoiding surgery and general anesthesia.
There are no approved drugs for children with sickle cell disease, but hydroxyurea is approved for the treatment of sickle cell disease in adults. In a meta-analysis, Strouse et al studied the efficacy, effectiveness, and toxicity of hydroxyurea in children with sickle cell disease. The investigators found that hemoglobin levels increased from 5-10% to 15-20%; hemoglobin concentration increased modestly (approximately 1 g/L) but significantly; hospitalizations decreased by 56-87%; and the frequency of pain crisis decreased. [22]
Radiography
The early plain radiographic findings of dactylitis consist of soft tissue swelling. Periosteal new-bone formation can be seen on radiographs 7-10 days later. Additionally, medullary expansion, cortical thinning, trabecular resorption, and resultant focal lucency may be seen 2-3 weeks after the onset of symptoms, but these findings usually resolve within weeks (see the images below).
Skeletal sickle cell anemia. Hand-foot syndrome. Soft tissue swelling with periosteal new-bone formation and a moth-eaten lytic process at the proximal aspect of the fourth phalanx.
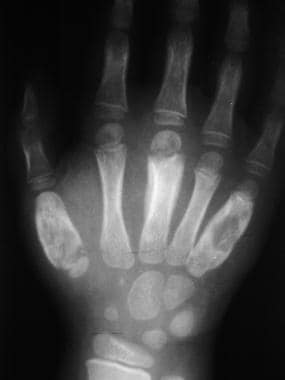 Skeletal sickle cell anemia. Advanced dactylitis. Lytic processes are present at the first and fifth metacarpals, along with periostitis, which is most prominent in the third metacarpal.
Skeletal sickle cell anemia. Advanced dactylitis. Lytic processes are present at the first and fifth metacarpals, along with periostitis, which is most prominent in the third metacarpal.
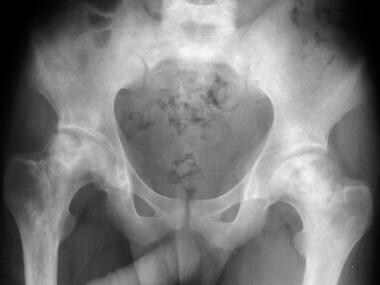 Skeletal sickle cell anemia. Osteonecrosis. Image shows flattening of the femoral heads with a mixture of sclerosis and lucency characteristic of osteonecrosis.
Skeletal sickle cell anemia. Osteonecrosis. Image shows flattening of the femoral heads with a mixture of sclerosis and lucency characteristic of osteonecrosis.
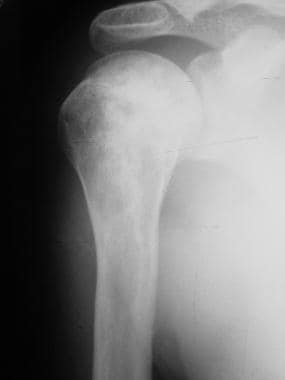 Skeletal sickle cell anemia. Bone infarct. Image shows patchy sclerosis of the humeral head and shaft representing multiple prior bone infarcts.
Skeletal sickle cell anemia. Medullary sclerosis. Image shows patchy sclerosis of the proximal tibia due to old infarctions. In other cases, sclerosis may be diffuse rather than patchy.
Skeletal sickle cell anemia. Bone infarct. Image shows patchy sclerosis of the humeral head and shaft representing multiple prior bone infarcts.
Skeletal sickle cell anemia. Medullary sclerosis. Image shows patchy sclerosis of the proximal tibia due to old infarctions. In other cases, sclerosis may be diffuse rather than patchy.
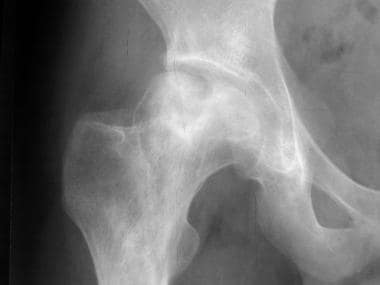
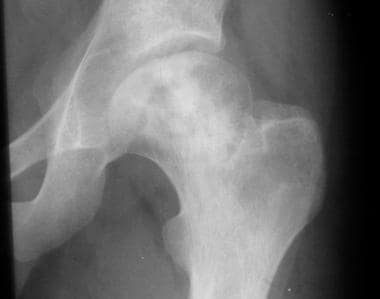
Osteonecrosis can affect the articular portions of the long bones. Collapse of the bone surfaces that are affected by osteonecrosis usually result in degenerative joint destruction. Radiographs of the affected joint may show patchy sclerosis initially, followed by flattening of bone (see the images below).
Widened medullary cavities may be noted. In the calvaria, the major trabecular spicules in the widened diploic space are aligned perpendicular to the inner table, giving the skull the characteristic hair-on-end appearance (see the images below).
Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
Skeletal sickle cell anemia. Detailed view of the expanded medullary cavity in the same patient as in the previous image.
Thickened bone can be observed. In the extremities, the medullary cavities are widened and the cortices are thinned, with loss of the normal modeling of the bone due to marrow hyperplasia.
Infarction of the central portion of the vertebral endplates results in the characteristic H deformity (angular depression) of vertebral bodies. When present in multiple vertebrae, this finding is virtually pathognomonic for sickle cell disease (see the image below).
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
Shortening of bone may be depicted. Epiphyseal infarction may produce cone-shaped epiphysis or premature fusion of the epiphysis resulting in abnormal shortening of the involved bone (see the image below).
Skeletal sickle cell anemia. Bone deformity. Image shows shortening of the second and third metacarpals and phalanges with partial or complete early fusion of the growth plates due to osteonecrosis in infancy. Osteomyelitis is now superimposed the third metacarpal.
After multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex (see the image below).
Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
Regarding myelosclerosis, the cumulative effects from repeated chronic small episodes of infarction result in a mottled, strandlike increased opacity in the medullary region (see the image below).
Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
Although radiography is not as sensitive as other studies for osteomyelitis in the first 1-2 weeks, plain images subsequently show cortical destruction, periosteal new bone, and (with time) sinus tracts and sequestra (see the images below).
Skeletal sickle cell anemia. Bone deformity. Image shows shortening of the second and third metacarpals and phalanges with partial or complete early fusion of the growth plates due to osteonecrosis in infancy. Osteomyelitis is now superimposed the third metacarpal.
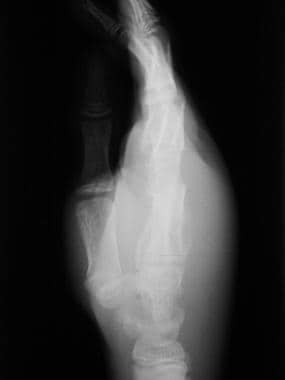 Skeletal sickle cell anemia. Radiograph of osteomyelitis shows a lytic process with periostitis and marked soft tissue swelling that is best seen on the lateral view.
Skeletal sickle cell anemia. Radiograph of osteomyelitis shows a lytic process with periostitis and marked soft tissue swelling that is best seen on the lateral view.
Computed Tomography
Although CT is not an initial study in most patients, CT scans can show findings of osteonecrosis, including sclerosis, collapse of bone (especially femoral heads), and a bone-within-bone appearance. CT may be useful to demonstrate subtle regions of osteonecrosis not apparent on plain radiographs in patients who are unable to have an MRI. [23]
Findings of osteomyelitis, including periosteal reaction, cortical or bone destruction, cloacae (sinus tracts), and sequestra or dead bone can be identified on CT scans (see the images below). Bone and soft tissue abscesses are demonstrated on contrast enhanced CT scans as low attenuation fluid collections with peripheral rim enhancement with or without internal gas. CT is not the test of choice for evaluation of acute osteomyelitis.
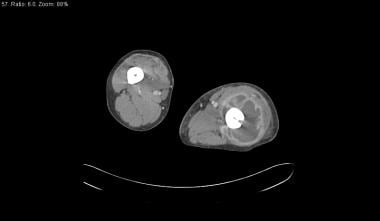 Skeletal sickle cell anemia. Osteomyelitis. CT scan in a soft tissue window demonstrates a large abscess in the left thigh encircling the femur, with hypoattenuating pus surrounded by a rim of vivid enhancement.
Skeletal sickle cell anemia. Osteomyelitis. CT scan in a soft tissue window demonstrates a large abscess in the left thigh encircling the femur, with hypoattenuating pus surrounded by a rim of vivid enhancement.
 Skeletal sickle cell anemia. Osteomyelitis and bone-within-bone. Bone-window CT scan in the same patient as in the previous image shows a bone-within-bone appearance (concentric rings of cortical bone) in the right femur. On the left, a sinus tract (cloaca) traverses the lateral aspect of the femoral cortex, and a small, shardlike sequestrum is present deep to the sinus tract.
Skeletal sickle cell anemia. Osteomyelitis and bone-within-bone. Bone-window CT scan in the same patient as in the previous image shows a bone-within-bone appearance (concentric rings of cortical bone) in the right femur. On the left, a sinus tract (cloaca) traverses the lateral aspect of the femoral cortex, and a small, shardlike sequestrum is present deep to the sinus tract.
Magnetic Resonance Imaging
MRI allows the early detection of changes in bone marrow due to acute and chronic bone marrow infarction, marrow hyperplasia, osteomyelitis, and osteonecrosis. As with plain radiography, the sine qua non of diagnosing osteomyelitis on MRI is the identification of cortical destruction for which MRI is exquisitely sensitive. [19, 19, 5, 20, 9]
Differentiation of bone infarct from osteomyelitis is one of the most challenging issues in the evaluation of acute bone pain in patients with SCD. Atypical findings on diffusion-weighted images have been identified in the early phase of presentation and can help differentiate bone infarct from osteomyelitis. [11]
Acute infarction is characterized by diffuse decreased signal intensity on T1-weighted images and increased signal intensity on T2-weighted images. This change results from bone marrow edema. With time, the process becomes focal, and T1-weighted images show a serpentine line of low signal intensity surrounding hyperintense marrow (see the images below). A double line of low signal intensity surrounding inner high signal intensity is sometimes seen on T2-weighted images.
Bone marrow hyperplasia in patients with sickle cell disease presents as diffuse areas of intermediate signal intensity on T1-weighted images, similar or slightly higher than that of skeletal muscle, and corresponding mildly increased signal on T2-weighted images. This effect is due to the replacement of fatty marrow by hematopoietic marrow.
Chronic bone infarcts, or old infarction and fibrosis, appears as focal areas of decreased signal intensity on both T1- and T2-weighted images. [21] With osteomyelitis, areas of bone infection demonstrate low signal intensity that replaces the usual high signal intensity of fatty marrow on T1-weighted images. This appears as areas of increased signal intensity on T2-weighted images and results from bone marrow edema and/or bone destruction. Bone sequestra, sinus tracts, and subperiosteal abscesses are also clearly identified when present. Fat saturated T1-weighted images with intravenous gadolinium facilitate the identification of bone sequestra, which present as focal low signal intensity defects surrounded by avidly enhancing inflammatory tissue. [9]
Skeletal sickle cell anemia. Osteonecrosis. Coronal T1-weighted MRI shows a slightly flattened femoral head with a serpentine margin of low signal intensity around an area of ischemic marrow with signal intensity similar to that of fat.
Skeletal sickle cell anemia. Osteonecrosis in the same patient as in the previous image. Coronal T2-weighted MRI shows a serpentine area of low signal intensity and additional focal areas of abnormal low signal intensity in the femoral head; these findings reflect collapse of bone and sclerosis.
Nuclear Imaging
Technetium-99m (99mTc) bone scanning can be used to detect early stages of osteonecrosis, and it is not as costly as MRI. Osteonecrosis can be detected on bone scans, appearing mostly as focal areas of increased activity. Occasionally, areas of decreased uptake can be seen; this is usually seen in early disease. Technetium-99m bone marrow scans demonstrate areas of decreased activity in marrow infarction. [24, 9]
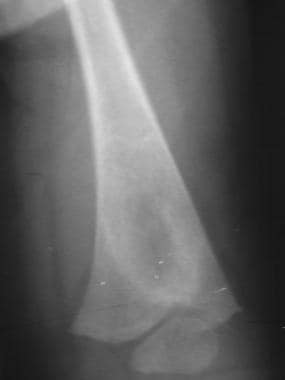 Skeletal sickle cell anemia. Bone infarction in an infant (see also next image). Image shows a curvilinear area of sclerosis with central lucency in the metaphysis of the femur representing a bone infarct.
Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.
Skeletal sickle cell anemia. Bone infarction in an infant (see also next image). Image shows a curvilinear area of sclerosis with central lucency in the metaphysis of the femur representing a bone infarct.
Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.
Indium-111 (111In) white blood cell (WBC) scanning is useful to diagnose osteomyelitis, which appears as an area of increased activity within bone. However, areas of marrow proliferation, which are common in patients with sickle cell, would also demonstrate increased activity on 111In WBC scans. The combination of a bone scan and a bone marrow scan has been used to differentiate acute osteomyelitis from bone infarcts in patient with sickle cell, since the clinical presentation of these 2 conditions may be very similar. Areas of bone infarction may be identified by decreased activity on the bone marrow scan with corresponding abnormal uptake on the bone scan. Acute osteomyelitis demonstrates increased activity on the bone scan, with normal activity on the bone marrow scan. [9, 25]
Single-photon emission computed tomography (SPECT)/CT has become an important tool to evaluate the different bone lesions in SCD, such as osteopenia, osteoporosis, avascular necrosis, and pathologic fracture. [15]
Questions & Answers
Overview
What is sickle cell disease (SCD)?
Which imaging modalities are used in the workup of sickle cell disease (SCD)?
What is the role of genetics in the etiology of sickle cell disease (SCD)?
What is the pathophysiology of sickle cell disease (SCD)?
How is sickle cell disease (SCD) treated?
What is the role of radiography in the workup of sickle cell disease (SCD)?
What is the role of CT scanning in the workup of sickle cell disease (SCD)?
What is the role of MRI in the workup of sickle cell disease (SCD)?
What is the role of nuclear imaging in the workup of sickle cell disease (SCD)?
-
Skeletal sickle cell anemia. H vertebrae. Lateral view of the spine shows angular depression of the central portion of each upper and lower endplate.
-
Skeletal sickle cell anemia. Hand-foot syndrome. Soft tissue swelling with periosteal new-bone formation and a moth-eaten lytic process at the proximal aspect of the fourth phalanx.
-
Skeletal sickle cell anemia. Advanced dactylitis. Lytic processes are present at the first and fifth metacarpals, along with periostitis, which is most prominent in the third metacarpal.
-
Skeletal sickle cell anemia. Expanded medullary cavity. The diploic space is markedly widened due to marrow hyperplasia. Trabeculae are oriented perpendicular to the inner table, giving a hair-on-end appearance.
-
Skeletal sickle cell anemia. Detailed view of the expanded medullary cavity in the same patient as in the previous image.
-
Skeletal sickle cell anemia. Osteonecrosis. Image shows flattening of the femoral heads with a mixture of sclerosis and lucency characteristic of osteonecrosis.
-
Skeletal sickle cell anemia. Osteonecrosis. Detail of the right hip.
-
Skeletal sickle cell anemia. Osteonecrosis. Detail of the left hip.
-
Skeletal sickle cell anemia. Bone infarct. Image shows patchy sclerosis of the humeral head and shaft representing multiple prior bone infarcts.
-
Skeletal sickle cell anemia. Chronic infarcts and secondary osteoarthritis. Image shows advanced changes of irregular sclerosis and lucency on both sides of the knee joint reflecting numerous prior infarcts. The joint surfaces are irregular and the cartilages are narrowed due to secondary osteoarthritis.
-
Skeletal sickle cell anemia. Bone-within-bone appearance. Following multiple infarctions of the long bones, sclerosis may assume the appearance of a bone within a bone, reflecting the old cortex within the new cortex.
-
Skeletal sickle cell anemia. Medullary sclerosis. Image shows patchy sclerosis of the proximal tibia due to old infarctions. In other cases, sclerosis may be diffuse rather than patchy.
-
Skeletal sickle cell anemia. Osteonecrosis. Coronal T1-weighted MRI shows a slightly flattened femoral head with a serpentine margin of low signal intensity around an area of ischemic marrow with signal intensity similar to that of fat.
-
Skeletal sickle cell anemia. Osteonecrosis in the same patient as in the previous image. Coronal T2-weighted MRI shows a serpentine area of low signal intensity and additional focal areas of abnormal low signal intensity in the femoral head; these findings reflect collapse of bone and sclerosis.
-
Skeletal sickle cell anemia. Osteomyelitis. CT scan in a soft tissue window demonstrates a large abscess in the left thigh encircling the femur, with hypoattenuating pus surrounded by a rim of vivid enhancement.
-
Skeletal sickle cell anemia. Osteomyelitis and bone-within-bone. Bone-window CT scan in the same patient as in the previous image shows a bone-within-bone appearance (concentric rings of cortical bone) in the right femur. On the left, a sinus tract (cloaca) traverses the lateral aspect of the femoral cortex, and a small, shardlike sequestrum is present deep to the sinus tract.
-
Skeletal sickle cell anemia. Bone deformity. Image shows shortening of the second and third metacarpals and phalanges with partial or complete early fusion of the growth plates due to osteonecrosis in infancy. Osteomyelitis is now superimposed the third metacarpal.
-
Skeletal sickle cell anemia. Radiograph of osteomyelitis shows a lytic process with periostitis and marked soft tissue swelling that is best seen on the lateral view.
-
Skeletal sickle cell anemia. Osteomyelitis. Coronal T1-weighted MRI shows marrow edema in the shortened third metacarpal, which appears dark. Note the loss of cortex along the radial aspect of the metacarpal.
-
Skeletal sickle cell anemia. Osteomyelitis in the same patient as in the previous image. On this T2-weighted MRI, the marrow and surrounding tissues are very bright as a result of inflammatory edema.
-
Skeletal sickle cell anemia. Osteomyelitis in the same patient as in the previous image. Axial T2-weighted MRI again shows the marrow edema and surrounding subperiosteal pus collection, with a thin, dark rim representing the periosteal membrane. There is diffuse bright signal intensity in the soft tissues; this represents inflammation and edema.
-
Skeletal sickle cell anemia. Bone infarction in an infant (see also next image). Image shows a curvilinear area of sclerosis with central lucency in the metaphysis of the femur representing a bone infarct.
-
Skeletal sickle cell anemia. Bone scan of bone infarct shows an area of increased uptake in the distal femoral metaphysis corresponding to the infarct demonstrated on the previous plain radiograph.







