Practice Essentials
In 1912, a pathologist named Pick coined the term pheochromocytoma —after the Greek words phaios, meaning dark or dusky, and chroma, meaning color—to describe the chromaffin reaction seen in adrenomedullary tumors. The tumors arise from the chromaffin cells of the adrenal medulla and are associated with increased catecholamine production. Although chromaffin tissue is also present elsewhere in the body, such as in the mediastinum, along the aorta, and in the pelvis, the term pheochromocytoma is reserved for tumors that arise from the adrenal medulla. Chromaffin cell tumors at other locations are more appropriately called paragangliomas or chemodectomas, although the term extra-adrenal pheochromocytoma is still applied. [1, 2] (Examples of pheochromocytomas are shown below.)
Patients with sporadic pheochromocytoma present at a mean age of approximately 44 years, and those with a genetic predisposition present at about 25 years of age. [1] Annual incidence is reported to be 2-8 per million and prevalence 0.05–0.12%. [2]
According to the Society of Surgical Oncology (SSO) Endocrine and Head and Neck Disease Site Working Group, pheochromocytomas constitute about 4–8% of all adrenal incidentalomas, and approximately 21.1–57.6% of all pheochromocytomas are discovered incidentally on imaging studies. [3]
Symptomatic patients typically present in their fourth or fifth decade of life, and between 90 and 95% of the tumors are unifocal and benign. The likelihood of malignancy depends on the primary site (adrenal vs. extra-adrenal) and the presence of certain germline mutations, such as SDHB in particular. [3]
Imaging modalities
Patients who may be referred for imaging of the adrenal glands include those with new or worsening diabetes mellitus (owing to impaired glucose regulation) and those with hypertensive crisis after anesthesia, surgery, or treatment with medications. Imaging may also be performed in patients with a known history of multiple endocrine problems. [4, 5, 6, 7, 8, 9, 10, 11]
The Endocrine Society recommends CT as the initial imaging study, but MRI is a better option in patients with metastatic disease or when radiation exposure must be limited. [12] MRI is more specific for pheochromocytomas than CT scanning, but MRI is not tolerable for some patients.
Computed tomography (CT) scanning and magnetic resonance imaging (MRI) have higher sensitivity in detecting pheochromocytomas than nuclear medicine scanning with iodine-131 metaiodobenzylguanidine (131I-MIBG), although 131I-MIBG uptake is more specific. Some authors prefer to use MIBG uptake scanning as the initial screening modality because it enables whole-body imaging, making it useful for the detection of extra-adrenal tumors and metastatic deposits. [11, 1, 13, 14, 15, 2, 16, 17, 18, 19, 20]
Positron emission tomography (PET) is also an alternative imaging test, with agents such as 18F-deoxyglucose (F-FDG), 18F-dihydroxyphenaline (F-DOPA), and 18F-fluorodopamine (F-FDA) being used in cases of negative 123I-MIBG when there is a high clinical and laboratory suspicion. PET with 18F-FDG has been shown to have a greater sensitivity than 123I-MIBG scintigraphy for metastatic disease because, in these cases, the tumors are generally less differentiated and therefore lose the ability to efficiently capture 123I-MIBG. [21]
Once an adrenal or extra-adrenal tumor is detected, CT scanning or MRI of the region may be performed for anatomic localization prior to surgical removal. If 131I-MIBG uptake is negative but the clinical findings suggest pheochromocytoma, CT scanning or MRI of the chest or abdomen may be performed, because the false-negative rate of MIBG scintigraphy is 10%. [22, 1, 2, 17, 18, 19, 20]
Luster et al investigated the specificity and sensitivity of (18)F-3,4-dihydroxyphenylalanine (DOPA) positron emission tomography (PET) scanning, CT scanning, and a combination of the 2 modalities in the identification and localization of adrenal and extra-adrenal pheochromocytomas. Nineteen lesions were detected by all 3 imaging methods, but only DOPA PET/CT scanning accurately characterized and localized them all, demonstrating, on a per-patient basis, a sensitivity of 100% and a specificity of 88%. [23]
A further study by Gaertner et al concluded the (18)F-LMI1195 is a promising tracer for tumor imaging. [24] Saad et al found in a study of 23 patients with a history of pheochromocytoma/paraganglioma that (18)F-FDG PET/CT was a superior tool for the localization of recurrent tumors. [25]
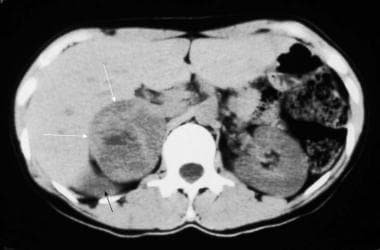 Nonenhanced computed tomography (CT) scan in a 35-year-old woman with hypertension demonstrates a large, right-sided, inhomogeneous adrenal mass (white arrows) with a central area of low attenuation that represents hemorrhage or necrosis. The upper pole of the displaced right kidney can be seen (black arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Nonenhanced computed tomography (CT) scan in a 35-year-old woman with hypertension demonstrates a large, right-sided, inhomogeneous adrenal mass (white arrows) with a central area of low attenuation that represents hemorrhage or necrosis. The upper pole of the displaced right kidney can be seen (black arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
 Axial gradient-recalled magnetic resonance angiogram in a 42-year-old woman with a 5-year history of hypertension who underwent magnetic resonance angiography for the assessment of renal arterial stenosis. Although the renal arteries were unremarkable, a 7.5-cm X 5-cm right adrenal mass was incidentally identified. Angiogram demonstrates a large, right-sided, inhomogeneous adrenal mass (arrows). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Axial gradient-recalled magnetic resonance angiogram in a 42-year-old woman with a 5-year history of hypertension who underwent magnetic resonance angiography for the assessment of renal arterial stenosis. Although the renal arteries were unremarkable, a 7.5-cm X 5-cm right adrenal mass was incidentally identified. Angiogram demonstrates a large, right-sided, inhomogeneous adrenal mass (arrows). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Detecting the tumors is important for a number of reasons. First, hypertension is usually cured with the removal of the tumor, whereas untreated patients are at risk for a lethal hypertensive paroxysm and long-term sequelae of the disease. Second, the discovery of a pheochromocytoma may indicate the presence of a familial disorder. Third, approximately 10% of pheochromocytomas are malignant. Incidentally, pheochromocytomas are called the 10% tumor because they are associated with a 10% risk of malignancy, because 10% of the tumors are bilateral, and because 10% of the tumors are extra-adrenal. Early detection may reduce the risk of metastasis.
Usually, tumors are larger than 3 cm when seen. They are highly vascular (see the images below), and larger tumors are prone to hemorrhage and necrosis, even when they are benign.
 Selective adrenal angiogram demonstrates the highly vascular nature of a pheochromocytoma. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Selective adrenal angiogram demonstrates the highly vascular nature of a pheochromocytoma. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
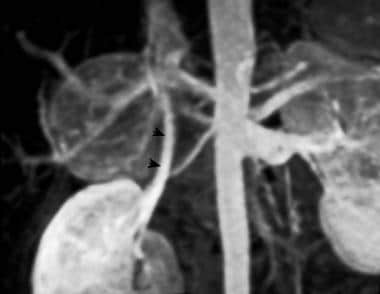 Three-dimensional maximal-intensity magnetic resonance angiogram clearly demonstrates a large, hypervascular right adrenal mass. Arrowheads demarcate the right renal vein. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Three-dimensional maximal-intensity magnetic resonance angiogram clearly demonstrates a large, hypervascular right adrenal mass. Arrowheads demarcate the right renal vein. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Failure to make the correct diagnosis can create serious risks for the patient. Early surgical removal of the adrenal gland is important to prevent complications associated with pheochromocytomas. Delay can significantly increase the risk of an adverse event. Worse, if pheochromocytoma is not considered in the diagnosis, the injection of contrast material, especially ionic contrast material, can provoke a hypertensive crisis.
Therefore, exclusion of pheochromocytoma should be part of the diagnostic evaluation in every patient with a suprarenal mass. Some authors recommend 131I-MIBG scintigraphy and/or the use of laboratory tests to confirm or rule out excessive production of catecholamines, prior to the use of invasive procedures.
Limitation of techniques
Unfortunately, the cost and lack of availability of MIBG studies restrict its use. In addition, imaging with 131I-MIBG can be time-consuming, and the technique has limited ability to provide sufficiently accurate information for surgery. Therefore, some authors recommend the use of at least 2 of the following modalities: CT scanning, MRI, and MIBG uptake studies. [1, 20]
CT scanning is quick and relatively inexpensive, and it offers good spatial localization. CT scan findings are not specific enough to distinguish masses caused by pheochromocytoma from other adrenal masses. Additionally, some authors report a risk of hypertensive crisis after the injection of contrast material.
MRI is more specific for pheochromocytomas than is CT scanning, but some patients cannot tolerate MRI.
Guidelines
The Endocrine Society recommends CT for initial imaging, but they note that MRI is a better option in patients with metastatic disease or when radiation exposure must be limited. They add that 123I-metaiodobenzylguanidine scintigraphy is a useful imaging modality for metastatic pheochromocytomas and paragangliomas (PPGL). [12]
The SSO Endocrine and Head and Neck Disease Site Working Group notes that CT is often the first imaging performed. MRI is not as commonly used as CT, and typically, MRI shows increased signal intensity on T2-weighted imaging and, as with CT, variable patterns of postcontrast enhancement. Historically, 123I-metaiodobenzylguanidine (MIBG) has been the primary functional imaging method, but a number of studies have consistently demonstrated the superiority of 68Ga-DOTATATE PET/CT over other functional imaging methods. [3]
The European Society of Endocrinology suggests screening for metastatic tumors by [18F]-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT) and performing imaging tests every 1-2 years in patients with biochemically inactive PPGLs to screen for local or metastatic recurrences or new tumors. [26]
The European Association of Nuclear Medicine (EANM) and Society of Nuclear Medicine and Molecular Imaging (SNMMI) published the following information regarding image acquisition and interpretation [27] :
-
PPGLs have different preferential sites of origin that must be known. Integration of functional and anatomic imaging in hybrid SPECT/CT or PET/CT devices is strongly recommended.
-
Images are generally acquired from the top of the skull (for a large jugular PGL) to the pelvis. In cases where recurrent or metastatic disease is suspected, whole-body images are necessary.
-
Metastases from PPGLs are often small and numerous and could be difficult to localize precisely on co-registered CT images obtained by combined SPECT/CT and PET/CT (plain CT, thick anatomical sections, or a positional shift between CT and nuclear images).
The following guidelines were provided by societies taking a multidisciplinary approach, including the Spanish Societies of Endocrinology and Nutrition (SEEN), Medical Oncology (SEOM), Medical Radiology (SERAM), Nuclear Medicine and Molecular Imaging (SEMNIM), Otorhinolaryngology (SEORL), Pathology (SEAP), Radiation Oncology (SEOR), and Surgery (AEC) and Spanish National Cancer Research Center (CNIO) [28] :
-
The diagnosis of PPGL relies on the imaging identification of an appropriately located mass with consistent clinical and biochemical features. Once the diagnosis is clinically and biochemically confirmed, imaging studies should be performed to localize and stage the tumor.
-
CT is the most common imaging method used because it is widely available and because it is less expensive and offers better spatial resolution than MRI.
-
MRI is not a first-choice imaging tool, but it has the advantage of being free of ionizing radiation and is suitable for children, pregnant women, and patients with adverse reactions to iodinated contrast medium.
-
PPGL cells express different transporters on their surface that allow images to be obtained by different radiotracers depending on the capture mechanisms, thus yielding different functional information. The most sensitive functional image for each tumor will depend on the clinical and biochemical profiles and the location of the primary tumor, which are also predictors of the underlying genotype.
-
Positron emission tomography (PET)/CT technology has been shown to be superior to scintigraphy with single photon emission computed tomography (SPECT/CT), with higher spatial resolution, greater sensitivity, and fewer indeterminate or equivocal studies.
-
Generally, when facing metastatic disease, better results are reported with the use of 18F-FDG PET/CT.
Radiography
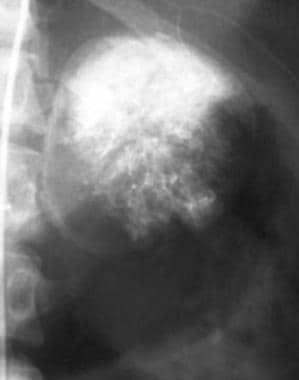
In comparison with other available modalities, radiography has limited value. Large adrenal masses may compress and deform the upper pole of the kidney (as shown in the image below); these may be discovered incidentally on intravenous urograms.
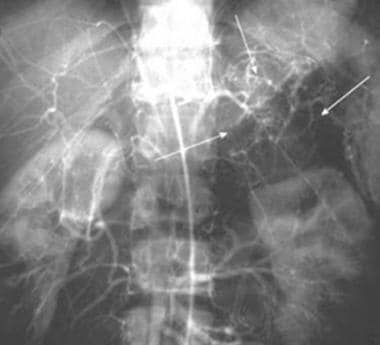 Abdominal aortogram in a patient with pheochromocytoma demonstrates a hypervascular mass (arrows) that flattens the upper pole of the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Abdominal aortogram in a patient with pheochromocytoma demonstrates a hypervascular mass (arrows) that flattens the upper pole of the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Computed Tomography
Pheochromocytomas are large tumors (often >3 cm), and they are usually round or oval masses with an attenuation similar to that of the liver. Larger lesions frequently demonstrate necrosis, hemorrhage, and fluid-fluid levels (see the images below). As a result, they often appear inhomogeneous. Calcification is rare, but it is reported. [16] CT scanning has a sensitivity of greater than 93% in the detection of pheochromocytomas and a specificity of 95% in the diagnosis of these tumors. [23, 29]
Pheochromocytomas can have a varied appearance on non-contrast CT, ranging from low-density to soft-tissue attenuation and from solid to complex or cystic. Most pheochromocytomas have an attenuation value greater than 10 HU. [1]
Nonenhanced computed tomography (CT) scan in a 35-year-old woman with hypertension demonstrates a large, right-sided, inhomogeneous adrenal mass (white arrows) with a central area of low attenuation that represents hemorrhage or necrosis. The upper pole of the displaced right kidney can be seen (black arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
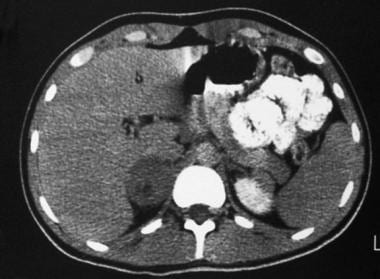 Late-phase contrast-enhanced computed tomography (CT) scan in a 22-year-old man with hypertension reveals an enlarged right adrenal gland with central necrosis. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Late-phase contrast-enhanced computed tomography (CT) scan in a 22-year-old man with hypertension reveals an enlarged right adrenal gland with central necrosis. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
As a result of the large size of the tumors, contrast material is not essential for their detection. In addition, some authors believe that the administration of the contrast agent may precipitate a hypertensive crisis in an unmedicated patient, although newer reports counter this view. Some authors recommend alpha-adrenergic and beta-adrenergic blockade prior to administration of contrast material. When administered, the tumor demonstrates varying degrees of enhancement. [20]
The adrenal gland on the right side is located directly posterior to the inferior vena cava (IVC), between the right crus of the diaphragm and the liver. The left adrenal gland is located more caudally and may be seen on the same imaging section that shows the kidney. On CT and axial MRI scans each gland is seen as a linear or inverted Y- or V-shaped organ located superior, medial, and anterior to each kidney. Note that neither CT scanning nor MRI can be used to distinguish between the adrenal cortex and the medulla.
Large, necrotic masses can be seen in other conditions, such as adrenal cortical carcinomas and metastasis. Thus, the diagnosis must be made in the setting of an appropriate clinical history. Patients with multiple endocrine neoplasia (MEN) syndromes may have atypical findings, such as thickened and nodular adrenal glands without large, discrete masses. [29]
Magnetic Resonance Imaging
The superior tissue characterization capability of MRI imaging combined with its multiplanar abilities affords it an advantage over CT scanning in the imaging of pheochromocytomas. [29] MRI is as sensitive as CT scanning, with sensitivities ranging from 86 to 100%. [29]
On MRI scans, pheochromocytomas are usually hypointense or isointense relative to the liver on T1-weighted spin-echo (SE) images, and they are highly intense on T2-weighted SE images. The reason for this difference is unknown, but it likely results from the high water content in cellular homogeneous tumors or from the high water content in necrotic regions. Tumors that have bled show the features typical of hemorrhage, depending on the age of the hemorrhage. T1- and T2-weighted MRI scans appear below. [1, 2]
The classic finding on T2-weighted MRI sequences is a marked hyperintensity known as a light-bulb bright lesion. [1]
(See the images below.)
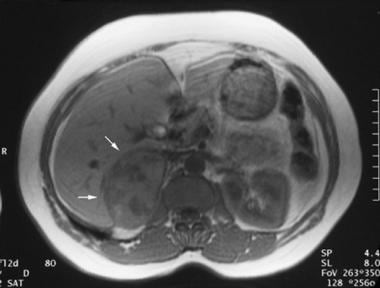
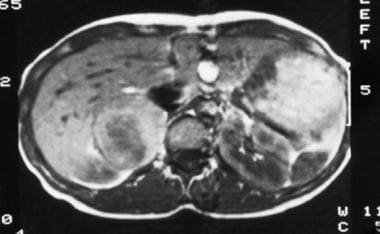 T1-weighted spin-echo magnetic resonance image (repetition time, 600 milliseconds; echo time, 15 milliseconds) in a 35-year-old woman with hypertension reveals a mixed isointense-to-hypointense right adrenal mass. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
T1-weighted spin-echo magnetic resonance image (repetition time, 600 milliseconds; echo time, 15 milliseconds) in a 35-year-old woman with hypertension reveals a mixed isointense-to-hypointense right adrenal mass. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
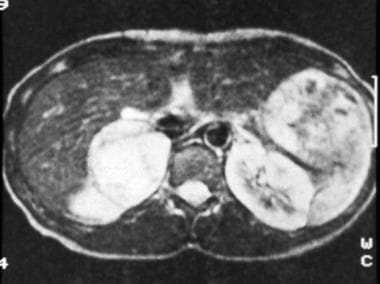 T2-weighted spin-echo magnetic resonance image (repetition time, 2000 milliseconds; echo time, 70 milliseconds) in a 35-year-old woman with hypertension (same patient as in the previous image) shows that the right adrenal tumor has high signal intensity, a feature typical of pheochromocytomas. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
T2-weighted spin-echo magnetic resonance image (repetition time, 2000 milliseconds; echo time, 70 milliseconds) in a 35-year-old woman with hypertension (same patient as in the previous image) shows that the right adrenal tumor has high signal intensity, a feature typical of pheochromocytomas. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
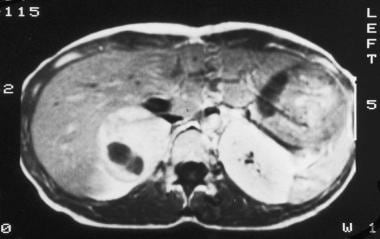 T1-weighted gadolinium-enhanced magnetic resonance image (same patient as in the previous 2 images) shows a diffusely enhancing mass with a central area devoid of enhancement. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
T1-weighted gadolinium-enhanced magnetic resonance image (same patient as in the previous 2 images) shows a diffusely enhancing mass with a central area devoid of enhancement. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
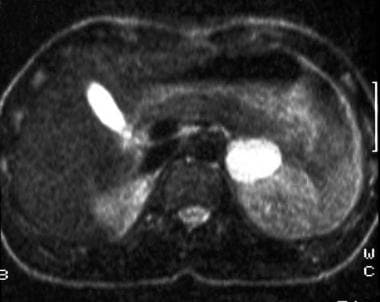 T2-weighted spin-echo magnetic resonance image (repetition time, 1800 milliseconds; echo time, 90 milliseconds) in a 30-year-old man with hypertension reveals a hyperintense mass in the left adrenal gland that posteriorly displaces the kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
T2-weighted spin-echo magnetic resonance image (repetition time, 1800 milliseconds; echo time, 90 milliseconds) in a 30-year-old man with hypertension reveals a hyperintense mass in the left adrenal gland that posteriorly displaces the kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
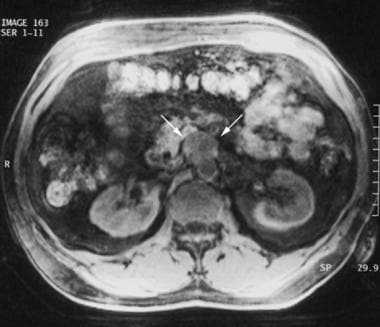 A 45-year-old patient with a history of multiple endocrine neoplasia type 2A who underwent prior thyroidectomy and bilateral adrenalectomy 14 years previously was admitted to the hospital with elevated blood pressure. Performance of breath-hold T1-weighted fat-suppressed gradient-recalled echo magnetic resonance imaging (MRI) (repetition time, 4.5 milliseconds; echo time, 1.9 milliseconds; flip angle, 12°) revealed an inhomogeneous, hypointense mass (arrows) anterior to the aorta. Later, at surgery, the tumor was proven to be a recurrent extra-adrenal pheochromocytoma (paraganglioma). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
A 45-year-old patient with a history of multiple endocrine neoplasia type 2A who underwent prior thyroidectomy and bilateral adrenalectomy 14 years previously was admitted to the hospital with elevated blood pressure. Performance of breath-hold T1-weighted fat-suppressed gradient-recalled echo magnetic resonance imaging (MRI) (repetition time, 4.5 milliseconds; echo time, 1.9 milliseconds; flip angle, 12°) revealed an inhomogeneous, hypointense mass (arrows) anterior to the aorta. Later, at surgery, the tumor was proven to be a recurrent extra-adrenal pheochromocytoma (paraganglioma). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
The use of flow-sensitive sequences is helpful in demonstrating the presence of intracaval extension of the tumor. On MRI scans obtained with gadolinium diethylenetriamine pentaacetic acid (DTPA), tumors demonstrate brisk and prolonged enhancement; however, contrast enhancement rarely provides additional information.
Gadolinium-based contrast agents have been linked to the development of nephrogenic systemic fibrosis (NSF) or nephrogenic fibrosing dermopathy (NFD). The disease has occurred in patients with moderate to end-stage renal disease after being given a gadolinium-based contrast agent to enhance MRI or magnetic resonance angiography (MRA) scans. Characteristics include red or dark patches on the skin; burning, itching, swelling, hardening, and tightening of the skin; yellow spots on the whites of the eyes ;joint stiffness with trouble moving or straightening the arms, hands, legs, or feet; pain deep in the hip bones or ribs; and muscle weakness.
Although pheochromocytomas typically have high signal intensity on T2-weighted images, this finding is not universal. In 20-33% of patients, T2-weighted images show atypical findings. As a result, an alternate diagnosis of necrotic metastasis or adrenal cortical carcinomas may be made if this variance is not kept in mind. On the other hand, comparable high signal intensity may be seen in some necrotic adrenal metastases and adrenal cysts; as a result, these lesions cannot always be distinguished from pheochromocytomas on MRI scans.
Ultrasonography
Ultrasonography has largely been replaced by CT scanning and MRI, and it is limited as a result of the effects of overlying bowel gas, especially in the assessment of the left adrenal gland. Therefore, the use of ultrasonography is limited to differentiating cystic lesions from solid lesions in the adrenal gland. Even in the pediatric population, MRI is the preferred imaging modality. (Examples of an ultrasonographically identified adrenal mass are seen below.)
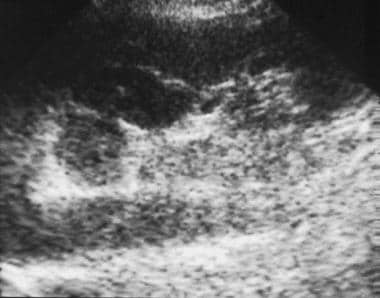 Ultrasonogram of an adrenal mass. Oblique sagittal image of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Ultrasonogram of an adrenal mass. Oblique sagittal image of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
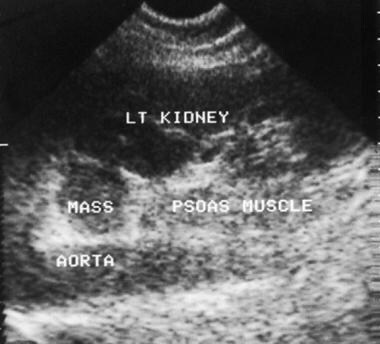 Ultrasonogram of an adrenal mass. Oblique sagittal image (same patient as in the previous image) of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Ultrasonogram of an adrenal mass. Oblique sagittal image (same patient as in the previous image) of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Nuclear Imaging
A normal adrenal medulla is seen in approximately 30% of patients, with an uptake of less than that of the liver. In pheochromocytoma, iodine-131 metaiodobenzylguanidine (131I-MIBG) scans show the tumor as a focal area in the adrenal gland that has prolonged increased uptake. (Such a focal area is seen in the image below.) Tumor metastases can be demonstrated in a similar fashion. Compared with 131I-MIBG imaging, 123I-MIBG imaging offers better image quality, single-photon emission CT (SPECT) capability, lower radiation exposure, and shorter imaging time. Reportedly, sensitivity is 86-90% for pheochromocytomas (especially in extra-abdominal tumors); specificity is as high as 99% with 131I-MIBG and is higher with 123I-MIBG (90% sensitivity, 100% specificity). [9, 10]
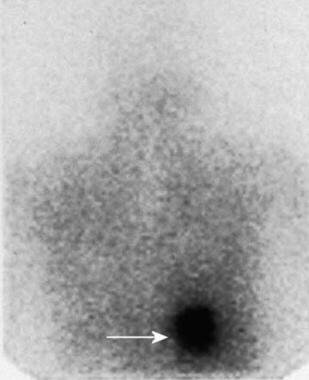 Patient admitted to the hospital with hypertensive crisis. Confirmatory iodine-131 metaiodobenzylguanidine scanning had been performed. A posterior 24-hour scan reveals a focus of increased tracer uptake in the region of the right adrenal gland. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
Patient admitted to the hospital with hypertensive crisis. Confirmatory iodine-131 metaiodobenzylguanidine scanning had been performed. A posterior 24-hour scan reveals a focus of increased tracer uptake in the region of the right adrenal gland. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
In a prospective, observational study, Timmers et al concluded that combined (18)F-fluorodopamine (FDA) PET/CT scanning is the best means of localizing primary pheochromocytomas and ruling out metastases (78% sensitivity for nonmetastatic pheochromocytomas, 76% sensitivity for metastatic pheochromocytomas), with 123I-MIBG scintigraphy and DOPA PET scanning being the second-best alternatives. The study included 20 patients with nonmetastatic pheochromocytomas (11 with adrenal pheochromocytomas), 28 patients with metastatic pheochromocytomas (13 with adrenal pheochromocytomas), and 4 patients in whom the presence of pheochromocytomas had been ruled out. [22]
Iodine-131 MIBG and 123I-MIBG are concentrated in the sympathomedullary system and then sequestered in neurosecretory granules. After pretreatment with Lugol iodine to saturate thyroid uptake, 0.5-1.0 mCi of 131I-MIBG or 9-10 mCi of 123I-MIBG is intravenously injected, and posterior adrenal images are obtained after 24, 48, and 72 hours. Technetium-99m DTPA also is used to improve localization of the kidneys.
Other nuclear imaging modalities include imaging with the somatostatin analogue octreotide and imaging with positron emitters, such as carbon-11 (11C) hydroxyephedrine, 2-[fluorine-18]fluoro-2-deoxy-D-glucose (FDG), and 11C epinephrine. [1]
The use of FDG, a glucose analogue used by metabolically active cells, with PET scanning is described. In a study of 29 patients with benign and malignant pheochromocytomas, Shulkin and colleagues reported tumoral uptake of FDG in 22 patients. [30] They noted that as many as 17 of the 29 patients had malignant pheochromocytoma, which may have resulted in this high degree of positivity.
Although the sensitivity and specificity of FDG PET were lower than those of MIBG scanning, FDG uptake occurred in all cases in which MIBG accumulation did not. Thus, when findings with other modalities fail to reveal or confirm the presence of the tumor, FDG PET may be useful. Other reports have since described the uptake of FDG in calvarial metastases from pheochromocytoma. [31, 32, 33, 17]
Studies of radionuclide imaging of sporadic pheochromocytoma have recommended 18F-FDOPA (fluorodopa) as the radiotracer of choice, with 68Ga-DOTATATE and FDG being considered second and third-line agents, respectively. [5]
False positives/negatives
MIBG uptake may be poorly visualized in tumors, even large tumors, with extensive necrosis. Occasionally, activity in the bowel can create false-positive findings, especially when extra-adrenal tumors are considered. The study can be repeated 24 hours later, when activity in the gut is displaced.
As a result of the 10% false-negative rate with MIBG scanning, some authors recommend abdominal CT scanning or MRI if a high clinical suspicion of pheochromocytoma exists but a causative tumor is not identified by assessing MIBG uptake.
-
Nonenhanced computed tomography (CT) scan in a 35-year-old woman with hypertension demonstrates a large, right-sided, inhomogeneous adrenal mass (white arrows) with a central area of low attenuation that represents hemorrhage or necrosis. The upper pole of the displaced right kidney can be seen (black arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
T1-weighted spin-echo magnetic resonance image (repetition time, 600 milliseconds; echo time, 15 milliseconds) in a 35-year-old woman with hypertension reveals a mixed isointense-to-hypointense right adrenal mass. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
T2-weighted spin-echo magnetic resonance image (repetition time, 2000 milliseconds; echo time, 70 milliseconds) in a 35-year-old woman with hypertension (same patient as in the previous image) shows that the right adrenal tumor has high signal intensity, a feature typical of pheochromocytomas. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
T1-weighted gadolinium-enhanced magnetic resonance image (same patient as in the previous 2 images) shows a diffusely enhancing mass with a central area devoid of enhancement. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
T2-weighted spin-echo magnetic resonance image (repetition time, 1800 milliseconds; echo time, 90 milliseconds) in a 30-year-old man with hypertension reveals a hyperintense mass in the left adrenal gland that posteriorly displaces the kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Late-phase contrast-enhanced computed tomography (CT) scan in a 22-year-old man with hypertension reveals an enlarged right adrenal gland with central necrosis. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Abdominal aortogram in a patient with pheochromocytoma demonstrates a hypervascular mass (arrows) that flattens the upper pole of the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Selective adrenal angiogram demonstrates the highly vascular nature of a pheochromocytoma. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Axial gradient-recalled magnetic resonance angiogram in a 42-year-old woman with a 5-year history of hypertension who underwent magnetic resonance angiography for the assessment of renal arterial stenosis. Although the renal arteries were unremarkable, a 7.5-cm X 5-cm right adrenal mass was incidentally identified. Angiogram demonstrates a large, right-sided, inhomogeneous adrenal mass (arrows). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Three-dimensional maximal-intensity magnetic resonance angiogram clearly demonstrates a large, hypervascular right adrenal mass. Arrowheads demarcate the right renal vein. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Patient admitted to the hospital with hypertensive crisis. Confirmatory iodine-131 metaiodobenzylguanidine scanning had been performed. A posterior 24-hour scan reveals a focus of increased tracer uptake in the region of the right adrenal gland. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Ultrasonogram of an adrenal mass. Oblique sagittal image of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Ultrasonogram of an adrenal mass. Oblique sagittal image (same patient as in the previous image) of the abdomen demonstrates an isoechoic mass of the left adrenal gland that is anterolateral to the aorta and medial to the left kidney. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Recurrent malignant pheochromocytoma in a 70-year-old woman who underwent a right adrenalectomy for pheochromocytoma in 1975. A T2-weighted spin-echo magnetic resonance image obtained in 1994 demonstrates a centrally hyperintense right-sided paraspinal mass (arrow), which was excised surgically and was proven to be a pheochromocytoma. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Recurrent malignant pheochromocytoma in a 70-year-old woman who underwent right adrenalectomy for pheochromocytoma in 1975 (same patient as in the previous image). Follow-up magnetic resonance image obtained in 1998 reveals a recurrent mass in the aortocaval region, positioned above an atrophic right kidney. Inversion recovery image (repetition time, 5000 milliseconds; echo time, 76 milliseconds; inversion time, 150 milliseconds) demonstrates an aortocaval mass (arrow). A right nephrectomy and excision of the recurrent mass were performed. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Recurrent malignant pheochromocytoma in a 70-year-old woman who underwent right adrenalectomy for pheochromocytoma in 1975 (same patient as in the previous 2 images). Follow-up sagittal inversion recovery fat-suppressed magnetic resonance image (repetition time, 5500 milliseconds; echo time, 76 milliseconds; inversion time 150 milliseconds) obtained in 1999 demonstrates an additional bilobed, hyperintense mass in the left periaortic region, which was believed to represent recurrence. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Recurrent malignant pheochromocytoma in a 70-year-old woman who underwent right adrenalectomy for pheochromocytoma in 1975 (same patient as in the previous 3 images). Follow-up computed tomography (CT) scans obtained in 2000 and a metaiodobenzylguanidine uptake scan (not shown) demonstrate a mass adjacent to the left renal vein (arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
A 45-year-old patient with a history of multiple endocrine neoplasia type 2A who underwent prior thyroidectomy and bilateral adrenalectomy 14 years previously was admitted to the hospital with elevated blood pressure. Performance of breath-hold T1-weighted fat-suppressed gradient-recalled echo magnetic resonance imaging (MRI) (repetition time, 4.5 milliseconds; echo time, 1.9 milliseconds; flip angle, 12°) revealed an inhomogeneous, hypointense mass (arrows) anterior to the aorta. Later, at surgery, the tumor was proven to be a recurrent extra-adrenal pheochromocytoma (paraganglioma). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
A 45-year-old patient with a history of multiple endocrine neoplasia type 2A who underwent prior thyroidectomy and bilateral adrenalectomy 14 years previously (same patient as in the previous image) was admitted to the hospital with elevated blood pressure. A gadolinium-enhanced gradient-recalled echo magnetic resonance image (repetition time, 4.5 milliseconds; echo time, 1.9 milliseconds) demonstrates peripheral enhancement of the mass (arrow). Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.
-
Inversion recovery fat-suppressed magnetic resonance image (repetition time, 5500 milliseconds; echo time, 76 milliseconds; inversion time, 150 milliseconds) reveals a large para-aortic mass, which is hyperintense. Courtesy of Dr Ali Shirkhoda, William Beaumont Hospital, Michigan.






