Practice Essentials
Pilocytic astrocytoma is a benign tumor of childhood, often located in deep midline structures such as the brainstem and the cerebellum. Although its rate of malignancy is low, CT and MRI features of brain pilocytic astrocytoma may resemble those of more aggressive brain tumors, and misdiagnosis may occur when it demonstrates findings that overlap with the features of more aggressive brain tumors. Although its rate of malignancy is low, CT and MRI features of brain PA may resemble those of more aggressive brain tumors, and misdiagnosis is particularly easy when it demonstrates findings that overlap with the features of more aggressive brain tumors. [1, 2, 3, 4]
Juvenile pilocytic astrocytomas occur more often in children and young adults. They are the most common astrocytic tumors in children, accounting for 80-85% of cerebellar astrocytomas and 60% of optic gliomas. Juvenile pilocytic astrocytomas usually arise in the cerebellum, brainstem, hypothalamic region, or optic pathways, but they may occur in any area where astrocytes are present, including the cerebral hemispheres and the spinal cord. The most common site of occurrence of juvenile pilocytic astrocytoma is the cerebellum. [5]
These tumors are usually discrete, indolent lesions associated with cyst formation. The cysts may be unilocular or multilocular, with an associated tumor nodule. The most common presenting symptoms are associated with increased intracranial pressure resulting from mass effect or hydrocephalus. Symptoms may include headache, nausea, vomiting, irritability, ataxia, and visual complaints, depending on the site of occurrence. [5, 6, 7, 8, 9, 10, 11, 12]
Imaging modalities
The classic juvenile pilocytic astrocytoma arises in a cerebellar hemisphere; it is easily seen on computed tomography (CT) and magnetic resonance imaging (MRI) scans, as a well-circumscribed lesion with an associated macrocyst. The nodular portion of the lesion usually demonstrates homogeneous contrast enhancement. Calcification is present in 10% of juvenile pilocytic astrocytomas. Other low-grade gliomas are typically hypoattenuating or hypointense, poorly defined, nonenhancing lesions on CT and MRI scans. [13, 14, 15, 16]
The imaging features of juvenile pilocytic astrocytoma do not adhere to the rules established for adult gliomas. Images frequently depict heterogeneous areas of avid enhancement, and MR spectroscopy reveals low N-acetylaspartate concentrations and high choline and lactate levels. All of these findings, if detected in an adult patient, raise concern for high-grade glioma (HGG). One of the most useful clues to the diagnosis of pilocytic astrocytoma is increased diffusivity on apparent diffusion coefficient maps. [17]
The preferred examination is MRI (demonstrated in the images below). [18, 19, 20, 21, 22, 23, 24]
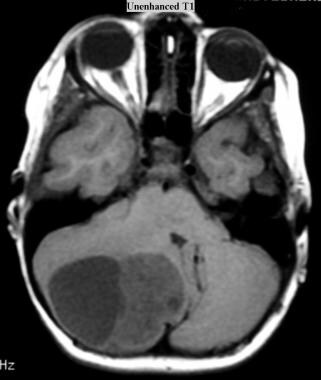 Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI without intravenous gadolinium contrast enhancement shows a cystic JPA in the right cerebellar hemisphere (same patient and tumor as in the next 2 images below).
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI without intravenous gadolinium contrast enhancement shows a cystic JPA in the right cerebellar hemisphere (same patient and tumor as in the next 2 images below).
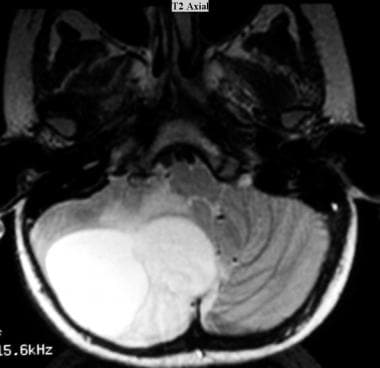 Juvenile pilocytic astrocytoma (JPA). Axial T2-weighted MRI shows a cystic JPA in the right cerebellar hemisphere. The fluid in the cyst has a higher signal intensity than that of the solid component. Peritumoral vasogenic edema is present.
Juvenile pilocytic astrocytoma (JPA). Axial T2-weighted MRI shows a cystic JPA in the right cerebellar hemisphere. The fluid in the cyst has a higher signal intensity than that of the solid component. Peritumoral vasogenic edema is present.
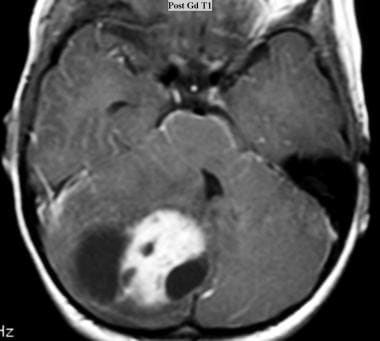 Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a cystic JPA with an enhancing component in the cyst in the right cerebellar hemisphere.
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a cystic JPA with an enhancing component in the cyst in the right cerebellar hemisphere.
Intervention
Pilocytic astrocytomas are typically treated with surgery. MRI scans are useful in outlining the contrast-enhancing tumor. The tumor should be completely resected whenever possible. Cyst wall enhancement may be seen on MRI scans; when such enhancement is present, resection of the entire cyst is indicated. [25, 26, 27, 28, 29, 24, 30] Use of radiologic findings alone to identify low-grade gliomas results in an incorrect diagnosis in as many as 50% of cases.
In patients in whom tumor resection is incomplete, the clinical course is often benign; postoperative stabilization of the disease is achieved, despite positive findings of tumor in surgical margins. For this reason, postoperative radiation therapy in these patients is controversial. Frequent follow-up with MRI is helpful.
Computed Tomography
Supratentorial juvenile pilocytic astrocytomas
Juvenile pilocytic astrocytomas may occur anywhere in the central nervous system. On CT scans, these astrocytomas cannot be reliably differentiated from other, more diffuse or aggressive tumors on the basis of imaging characteristics alone. CT scans may show hypoattenuating areas, isoattenuating areas, or both. Enhancement varies from none to extensive, with varying degrees of necrosis and cyst formation.
Supratentorial malignant glioma, ependymoma, and oligodendroglioma may have similar appearances. Lower-grade tumors tend to be homogeneous and well circumscribed. Peritumoral edema is mild, and no hemorrhage is present. Higher-grade tumors have more surrounding edema, are more heterogeneous in density, and may have areas of hemorrhage.
(A CT scan of a juvenile pilocytic astrocytoma is shown below.)
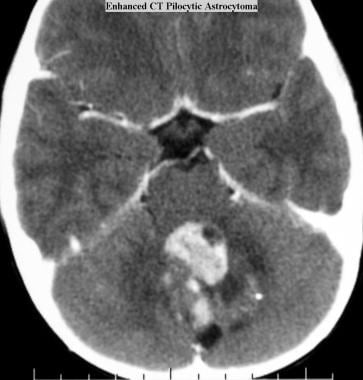 Juvenile pilocytic astrocytoma (JPA). Axial CT scan obtained with intravenous contrast material shows a contrast-enhancing JPA with cystic components in the cerebellum.
Juvenile pilocytic astrocytoma (JPA). Axial CT scan obtained with intravenous contrast material shows a contrast-enhancing JPA with cystic components in the cerebellum.
Optic nerve and optic chiasm hypothalamic juvenile pilocytic astrocytomas
A subset of astrocytic tumors occurs in patients with NF1. These tumors may involve the optic nerves, the optic chiasm, and the optic tracts. Most are juvenile pilocytic astrocytomas, but their imaging characteristics are not specific with regard to their histologic features. Varying degrees of cystic change and enhancement are demonstrated. These tumors may appear smooth, fusiform, eccentric, or lobulated. CT scanning demonstrates the intraorbital optic nerves and is sensitive in the detection of the tumors. About 20% of juvenile pilocytic astrocytomas have microscopic calcifications; these calcifications are less frequently seen on CT scans than on other types of images.
Posterior fossa juvenile pilocytic astrocytomas
Among pediatric tumors of the posterior fossa, astrocytomas are second in frequency only to medulloblastoma. Approximately 75% of cerebellar astrocytomas are of the pilocytic type; imaging does not help in predicting their histologic features because fibrillary forms may have similar appearances.
Imaging characteristics are most typical for cerebellar tumors during the first decade of life. The typical presentation of a juvenile pilocytic astrocytoma is of a large cerebellar hemispheric or vermian mass that is predominantly cystic in a child younger than 10 years. Nonenhanced CT scans show hypoattenuation or isoattenuation. Tumor contrast enhancement is homogeneous or heterogeneous, depending on the extent of the cystic necrotic changes.
Magnetic Resonance Imaging
Supratentorial juvenile pilocytic astrocytomas
On T1-weighted images, the signal intensity is generally low; on T2-weighted images, the signal intensity is increased. Enhancement patterns are similar to those depicted on CT scans.
(MRI images of juvenile pilocytic astrocytomas are shown below.)
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI without intravenous gadolinium contrast enhancement shows a cystic JPA in the right cerebellar hemisphere (same patient and tumor as in the next 2 images below).
Juvenile pilocytic astrocytoma (JPA). Axial T2-weighted MRI shows a cystic JPA in the right cerebellar hemisphere. The fluid in the cyst has a higher signal intensity than that of the solid component. Peritumoral vasogenic edema is present.
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a cystic JPA with an enhancing component in the cyst in the right cerebellar hemisphere.
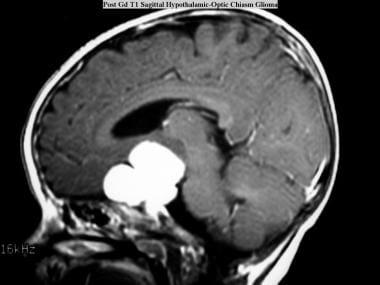 Juvenile pilocytic astrocytoma (JPA). Sagittal T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a JPA with enhancement in the hypothalamic area.
Juvenile pilocytic astrocytoma (JPA). Sagittal T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a JPA with enhancement in the hypothalamic area.
Optic nerve and optic chiasm hypothalamic juvenile pilocytic astrocytomas
Optic chiasm hypothalamic gliomas cannot be distinguished on the basis of their site of origin and are considered to be a single entity. On T1-weighted images, the signal intensity is low. On T2-weighted images, the signal intensity is generally increased. The increase in T2-weighted signal intensity may extend as far as the optic radiations, but such findings do not correlate directly with the presence of tumor. Enhancement is similar to that seen on CT scans. Use of fat-saturated T1-weighted postcontrast MRI of the intraorbital optic nerves is a sensitive method for demonstrating the tumor.
Posterior fossa juvenile pilocytic astrocytomas
The signal intensity is low with T1-weighted sequences and high with T2-weighted sequences. Enhancement patterns are similar to those seen on CT scans. MRI is less sensitive to calcium than is CT scanning.
Vermian tumors are often associated with hydrocephalus. Three general tumor patterns are found.
Less than 10% are solid. These tumors may enhance in a homogeneous or a heterogeneous fashion.
Approximately 50% are simple cysts with a single mural nodule. On CT and MRI scans, the nodule enhances homogeneously, but the associated cyst wall usually does not. Likewise, no histologic evidence of tumor is present in the cyst wall. For this kind of tumor, removal of the mural nodule may be sufficient for treatment.
About 40-45% consist of multilocular cysts. These are actually necrotic tumors; they have a cystlike appearance. The periphery or cyst wall enhances. Histologic evidence of tumor is present in the cyst wall. Cure requires resection of the entire wall. Imaging shows clear-cut enhancement of the nonnecrotic portions of the tumor.
Degree of confidence
Specific findings on MRI may be suggestive of juvenile pilocytic astrocytomas, but they are not diagnostic for this disease. Metastatic disease, neoplasm, and high-grade glioma cannot be excluded on the basis of radiographic findings. With regard to tumors of the posterior fossa, the most common possibilities in the differential diagnosis are medulloblastoma and ependymoma. Metastases are rare in childhood. Medulloblastomas are typically isoattenuating to hyperattenuating on nonenhanced CT scans.
Ependymomas may extend laterally or inferiorly to the foramina of Luschka or Magendie; extension is to the cerebellopontine angle or through the foramen magnum, respectively. They are isoattenuating to hyperattenuating on nonenhanced CT scans. About 50% of ependymomas exhibit small multifocal calcifications on CT scans. The major differential diagnostic consideration for optic chiasm/hypothalamic glioma is craniopharyngioma.
Gadolinium-based contrast agents have been linked to the development of nephrogenic systemic fibrosis (NSF) or nephrogenic fibrosing dermopathy (NFD). NSF/NFD has occurred in patients with moderate to end-stage renal disease after being given a gadolinium-based contrast agent to enhance MRI or MRA scans. NSF/NFD is a debilitating and sometimes fatal disease. Characteristics include red or dark patches on the skin; burning, itching, swelling, hardening, and tightening of the skin; yellow spots on the whites of the eyes; joint stiffness with trouble moving or straightening the arms, hands, legs, or feet; pain deep in the hip bones or ribs; and muscle weakness.
-
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI without intravenous gadolinium contrast enhancement shows a cystic JPA in the right cerebellar hemisphere (same patient and tumor as in the next 2 images below).
-
Juvenile pilocytic astrocytoma (JPA). Axial T2-weighted MRI shows a cystic JPA in the right cerebellar hemisphere. The fluid in the cyst has a higher signal intensity than that of the solid component. Peritumoral vasogenic edema is present.
-
Juvenile pilocytic astrocytoma (JPA). Axial T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a cystic JPA with an enhancing component in the cyst in the right cerebellar hemisphere.
-
Juvenile pilocytic astrocytoma (JPA). Sagittal T1-weighted MRI obtained with intravenous gadolinium-based contrast agent shows a JPA with enhancement in the hypothalamic area.
-
Juvenile pilocytic astrocytoma (JPA). Axial CT scan obtained with intravenous contrast material shows a contrast-enhancing JPA with cystic components in the cerebellum.








