Practice Essentials
Ganglioneuromas (GN) and ganglioneuroblastomas (GNB) are tumors of the sympathetic nervous system that originate from neural crest sympathogonia, which are completely undifferentiated cells of the sympathetic nervous system. Along with neuroblastomas, ganglioneuromas and ganglioneuroblastomas are collectively known as neuroblastic or neurogenic tumors. [1, 2]
Ganglioneuromas often present as a solitary, painless, slow-growing mass consisting of ganglion cells, Schwann cells, and fibrous tissue. The most commonly affected sites are the posterior mediastinum (41%), retroperitoneum (37%), adrenal gland (21%), and neck (8%). [3]
Ganglioneuroblastoma is a transitional tumor on the intermediate spectrum of disease between ganglioneuromas and neuroblastomas, containing elements of both malignant neuroblastoma and benign ganglioneuroma. [4] They are most common in children, with a median age at diagnosis of 22 months; most cases are diagnosed before 10 years of age. Although cases of adolescent or adult-onset GNB have been reported, they are extremely rare. [5]
These tumors occur most frequently in the abdomen, but they can grow wherever sympathetic nervous tissue is found. Common locations for ganglioneuromas and ganglioneuroblastomas include the adrenal gland, paraspinal retroperitoneum (sympathetic ganglia), posterior mediastinum, head, and neck; it is uncommon to find them in the urinary bladder, bowel wall, abdominal wall, and gallbladder. GNs are found incidentally in most cases and manifest as asymptomatic masses. The tumor could cause some complications if it becomes large enough to press against the adjacent organs. [6]
(See the images below.)
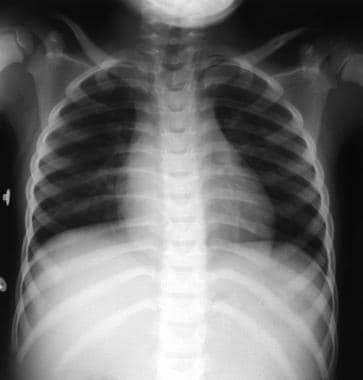 This frontal chest radiograph shows a right paraspinal well-defined density in the retrocardiac region. The right cardiophrenic angle appears normal.
This frontal chest radiograph shows a right paraspinal well-defined density in the retrocardiac region. The right cardiophrenic angle appears normal.
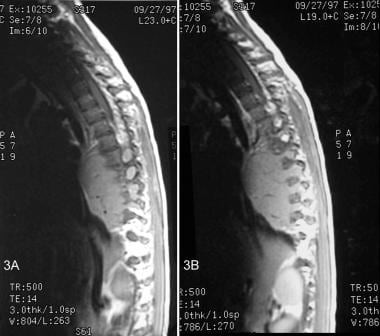 T1-weighted magnetic resonance images of a ganglioneuroma. The sagittal views demonstrate a well-defined, solid mass located slightly anterior to the midthoracic vertebral bodies. The mass extends into and widening multiple neural foramina. Flow voids within the lesion represents vascularity.
T1-weighted magnetic resonance images of a ganglioneuroma. The sagittal views demonstrate a well-defined, solid mass located slightly anterior to the midthoracic vertebral bodies. The mass extends into and widening multiple neural foramina. Flow voids within the lesion represents vascularity.
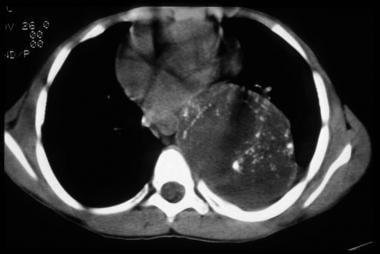 This axial noncontrast computed tomography image of a ganglioneuroblastoma demonstrates a large, left heterogeneous paraspinal lesion with speckled calcifications that are predominantly peripheral. The mass is displacing the mediastinum to the right.
This axial noncontrast computed tomography image of a ganglioneuroblastoma demonstrates a large, left heterogeneous paraspinal lesion with speckled calcifications that are predominantly peripheral. The mass is displacing the mediastinum to the right.
Preferred examination
Magnetic resonance imaging (MRI) and computed tomography (CT) scanning are the preferred methods for imaging ganglioneuromas and ganglioneuroblastomas. [7, 8, 9] MRI is the modality of choice for evaluating the extension of spinal tumors. [10, 11, 3, 12, 13, 14, 15]
CT scanning is the imaging modality that is most commonly used to evaluate neuroblastic tumors. It has proven to be the superior imaging technique when identifying tumor size, organ of origin, tissue invasion, vascular encasement, adenopathy, and calcifications. Newly diagnosed cases are evaluated with standard chest, abdominal, and pelvic CT scans. [10]
In general, neuroblastic or neurogenic tumors appear radiologically as well-circumscribed, smooth or lobulated masses that may contain calcifications. The benign (ganglioneuromas) and malignant (ganglioneuroblastomas) forms of these tumors are virtually identical radiologically. The only differentiating factor is the possibility of distant metastases with malignant ganglioneuroblastomas.
The International Neuroblastoma Staging System (INSS) was developed to stage these tumors by using clinical, radiologic, and surgical data. [10]
Complete surgical resection of ganglioneuromas is important, because it allows for good tissue sampling and a thorough pathology examination of the specimen to ensure a correct diagnosis of ganglioneuroma. In rare cases, these tumors recur; therefore, radiologic examination is an important tool for the proper localization and characterization of primary and recurrent tumors. These tumors are identified on radiologic examination on the basis of their location, shape, and internal structure; however, the diagnosis of ganglioneuroma cannot be based on radiologic findings alone. [16]
Radiography
Ganglioneuromas are typically discovered on a routine radiograph. However, on radiologic examination, all neuroblastic tumors (ie, ganglioneuroblastomas, ganglioneuromas, and neuroblastomas) look similar. The main difference is that ganglioneuroblastomas and neuroblastomas can possibility metastasize.
These tumors have highly variable radiologic appearances and growth behaviors that are reflective of their variable locations. Conventional radiographs show a retroperitoneal, posterior mediastinal, pelvis, or neck mass (see the following images). Up to 30% of these tumors may show calcifications. Rib and vertebral foraminal erosions, increased intercostal spaces, and vertebral body pedicle erosions may be seen with both retroperitoneal and posterior mediastinal tumors. Hepatomegaly, periostitis, widened cranial sutures, focal bone lucencies, focal bone sclerosis, and lucent submetaphyseal zones may indicate metastatic disease. [10]
This frontal chest radiograph shows a right paraspinal well-defined density in the retrocardiac region. The right cardiophrenic angle appears normal.
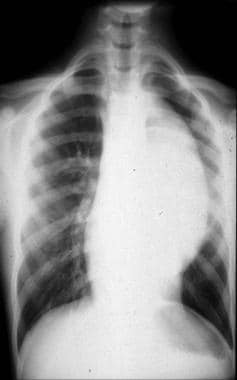 Frontal chest radiograph shows a large left paraspinal density. The lesion is silhouetting the aorta and, on the lateral view in the next image, appears to be retrocardiac and in close proximity to the vertebral column.
Frontal chest radiograph shows a large left paraspinal density. The lesion is silhouetting the aorta and, on the lateral view in the next image, appears to be retrocardiac and in close proximity to the vertebral column.
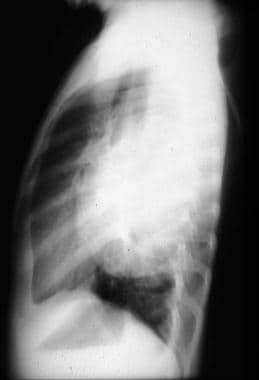 This lateral view radiograph of a ganglioneuroblastoma shows a large, well-defined homogeneous density that appears to be retrocardiac and in close proximity to the vertebral column.
This lateral view radiograph of a ganglioneuroblastoma shows a large, well-defined homogeneous density that appears to be retrocardiac and in close proximity to the vertebral column.
Computed Tomography
CT scanning is the imaging modality that is most commonly used to evaluate neuroblastic tumors. It has proven to be the superior imaging technique when identifying tumor size, organ of origin, tissue invasion, vascular encasement, adenopathy, and calcifications. Newly diagnosed cases are evaluated with standard chest, abdominal, and pelvic CT scans. [10]
Ganglioneuromas
Retroperitoneal and adrenal ganglioneuromas appear well-defined. Their shape ranges from round to lobulated, they show discrete and punctate calcifications in 42-60% of cases, and they tend to grow around major blood vessels. Ganglioneuromas do not compress these blood vessels, and blood flow remains normal. [1]
Nonenhanced CT scanning reveals a homogeneous mass with less attenuation than muscle. Ichikawa et al described a delayed heterogeneous uptake of contrast in a ganglioneuroma. [17] The reason for this type of uptake is that these tumors take longer to accumulate contrast material in the extracellular space. This delay is directly proportional to the amount of myxoid stroma in the tumor.
Ganglioneuroblastomas
Ganglioneuroblastomas have a variable appearance on CT scans and can be cystic or solid. [1] Abdominal and pelvic tumors are mostly heterogeneous and large; smaller tumors may appear homogeneous. (See the image below.)
This axial noncontrast computed tomography image of a ganglioneuroblastoma demonstrates a large, left heterogeneous paraspinal lesion with speckled calcifications that are predominantly peripheral. The mass is displacing the mediastinum to the right.
Areas of bleeding and necrosis may be as large as 4 cm and appear to be of lower attenuation. These tumors can grow around blood vessels but rarely invade them. Vessel compression, however, is a realistic possibility. If compression of renal vasculature occurs, hypertension can result. [10] Other vessels at risk include the splenic vein, inferior vena cava, aorta, celiac artery, and superior mesenteric artery (SMA).
Any metastasis to the liver or lung and adenopathy can also be identified on CT scans. Liver metastases appear as diffuse infiltration (in infants) and as focal areas of low enhancement. Metastatic disease of the lung can appear either as well-defined nodules or as larger areas of tissue consolidation. Although exceedingly rare, brain metastases do occur and have a variable appearance, ranging from hemorrhagic to cystic with rim-enhancement to a solid mass.
Intracranial dural involvement is a more common occurrence and can be seen as meningeal masses and meningeal enhancement. These metastatic tumors may be large enough to produce mass effect on the brain and cranium (suture widening). Metastatic involvement of the sphenoid bone that extends into the orbits produces a red and blue discoloration of the orbits that can be misinterpreted as physical abuse. [18]
Magnetic Resonance Imaging
MRI is a multiplanar imaging technique that creates images with better tissue discrimination than CT scanning. This makes MRI more effective at evaluating organs of origin, regional invasion, and intraspinal tumoral extension. Any patient with a paraspinal tumor should undergo an MRI to evaluate possible tumoral invasion of the epidural space. In these cases, coronal, sagittal, and axial views can demonstrate spinal cord and nerve root displacement, as well as superior and inferior tumor growth. [11, 14, 15]
(See the following images.)
T1-weighted magnetic resonance images of a ganglioneuroma. The sagittal views demonstrate a well-defined, solid mass located slightly anterior to the midthoracic vertebral bodies. The mass extends into and widening multiple neural foramina. Flow voids within the lesion represents vascularity.
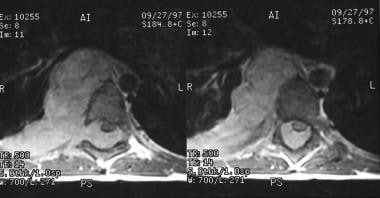 T1-weighted postcontrast axial images of a ganglioneuroma demonstrate homogeneous enhancement of the lesion. The lesion is extending from the right paraspinal region into the epidural space through the neural foramina causing cord compression.
T1-weighted postcontrast axial images of a ganglioneuroma demonstrate homogeneous enhancement of the lesion. The lesion is extending from the right paraspinal region into the epidural space through the neural foramina causing cord compression.
MRI is also useful for imaging bone marrow; the lesions appear hypointense on T1-weighted images and hyperintense on T2-weighted images. In addition, MRI can be used to detect hepatic metastases in infants with stage 4S disease, which can be missed on CT scanning because of its appearance as a uniform increase in liver attenuation. These lesions appear hyperintense on T2-weighted MRIs. [10, 19]
Ganglioneuromas
Ganglioneuromas appear homogeneous on MRIs and have a relatively intermediate signal intensity on all sequences. [20] Tumors with intermediate to high signal intensity on T2-weighted images have a higher degree of cellularity and more collagen. These tumors will have a small myxoid component. Markedly high T2 intensity, on the other hand, signifies a high myxoid stroma component and low cellularity and collagen amounts. [1]
Ganglioneuromas have characteristic curvilinear bands of low signal-intensity on T2-weighted images that impart a whorled pattern to the tumor. These bands are composed of collagen fibers and intertwined bundles of Schwann cells. The capsule surrounding the tumor appears as a low-density ring on both T1- and T2-weighted images.
Contrast enhancement with MRI is similar to contract enhancement with CT imaging. Early contrast enhancement occurs in areas of relatively high vascularity with high capillary permeability. However, delayed enhancement results when diffusion of the enhancing material into the extravascular space is slowed or impeded. [21] Therefore, fibrous tissues show slow or late enhancement. These tumors enhance gradually, with final enhancement ranging from none to marked. MRI is the modality of choice for evaluating the extension of spinal tumors. [10]
Ganglioneuroblastomas
Ganglioneuroblastomas appear heterogeneous on MRIs, with variable enhancement and low signal intensity on T1-weighted images and high signal intensity on T2-weighted images. [22] Tumor calcifications are more difficult to appreciate on MRIs than on CT scans; on MRIs, they appear as signal voids. Areas of hemorrhage have a high T1 signal intensity, and cystic areas have a high T2 signal intensity. [10] Ganglioneuroblastomas, like neuroblastomas and other malignant tumors, have a pattern of early enhancement followed by a partial washout; early enhancement is indicative of high vascularity. [21]
Differential diagnosis
The differential diagnosis of ganglioneuroma and ganglioneuroblastoma includes neuroblastoma, adrenal adenoma, adrenal carcinoma, neurofibroma, schwannoma, and pheochromocytoma. Ganglioneuromas can be differentiated from more aggressive neuroblastomas and ganglioneuroblastomas by their regular contours and lack of tissue invasion and vessel encasing, their occurrence in older patients, and their discrete, punctate calcifications on CT scans. Neuroblastomas and ganglioneuroblastomas tend to have amorphous or coarse calcifications. In addition, ganglioneuromas rarely metastasize, whereas neuroblastomas and ganglioneuroblastomas can metastasize to bone, skin, and other organs. [21]
It is more difficult to differentiate ganglioneuromas from schwannomas or neurofibromas. In general, schwannomas and neurofibromas are round and can cause bone erosion and destruction. Most ganglioneuromas are flat and elongated, and they normally do not affect bone. Schwannomas may demonstrate cystic degeneration that does not occur in ganglioneuromas. Neurofibromas do not have a capsule; the presence of one in a patient would point to a ganglioneuroma diagnosis. [21]
Adrenal disease can be differentiated from neuroblastic tumors by its unique enhancing patterns. Adrenal adenomas enhance and wash out early, whereas pheochromocytomas and adrenocortical carcinomas strongly enhance early but wash out at a slower rate. Adrenal carcinomas invade vascular structures in more than 50% of the cases. [21]
(MRIs of these tumors are provided below.)
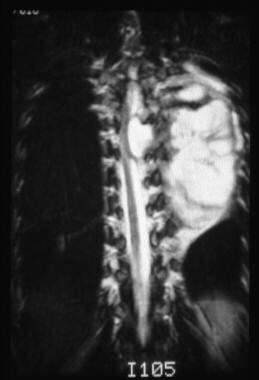 This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
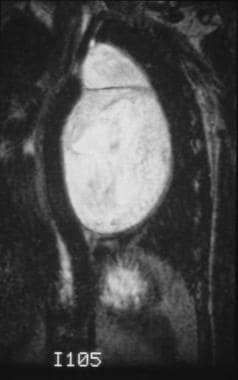 This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
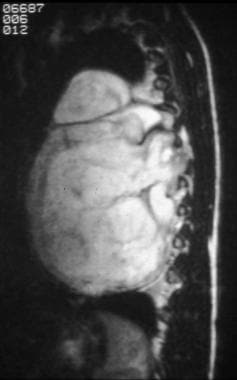 This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina.
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina.
Ultrasonography
Ultrasonography of a ganglioneuroma would show a homogeneous, hypoechoic, well-circumscribed mass. [19] Ganglioneuroblastomas show heterogeneic echogenicity, with areas of hemorrhage and necrosis appearing anechogeneic. Calcifications appear as either focal or diffuse areas of increased echogenicity with variable appearance of shadowing. Ultrasonography has proven to be useful in evaluating other organs for metastasis and for determining blood flow in blood vessels surrounded by tumor. [10]
Nuclear Imaging
In cases of ganglioneuroblastoma, scintigraphy is not only useful for identifying the primary tumor, but it is also useful for monitoring the pattern of metastatic spread. Primary and metastatic tumors take up catecholamine and somatostatin analogs.
Iodine-tagged metaiodobenzylguanidine (MIBG), a catecholamine analog, is used to identify catecholamine-producing tumors such as ganglioneuroblastoma, ganglioneuroma, neuroblastoma, pheochromocytoma, carcinoid tumor, and medullary thyroid cancer. [23, 24, 25, 26]
Technetium-99m (99mTc) testing is an initial study done in all patients with diagnosed ganglioneuroblastoma to determine the amount of metastatic disease present; however, as many as 75% of primary tumors also show uptake. This method is preferred for the examination of bone metastases, as cortical and marrow lesions are difficult to discriminate (as they are with MIBG studies). Metastases appear as areas of higher uptake, and they include areas such as the lung and liver. The amount of 99mTc uptake is, purportedly, directly related to calcium metabolism of the tumor cells.
Ganglioneuromas can accumulate MIBG in a fashion similar to ganglioneuroblastomas; tumors that uptake MIBG are catecholamine-producing tumors. [23, 24, 25, 26] Although 90-95% of ganglioneuroblastomas produce catecholamines, only approximately 70% take up MIBG. Therefore, negative results do not mean there is no disease. Despite this limitation, MIBG scintigraphy has an 88% sensitivity and a 99% specificity for tumors containing sympathetic tissue, such as ganglioneuroblastomas, ganglioneuromas, neuroblastomas, pheochromocytomas, and carcinoids. The disadvantage is that there is no way to discriminate the type of tumor in which the uptake occurs. When bone is affected, this method also makes staging difficult, because it does not distinguish cortical involvement from marrow involvement.
Unfortunately, up to 50% of recurrent tumors do not take up MIBG. This makes MIBG scintigraphy a poor method of posttherapeutic monitoring for recurrences. Somatostatin-analog testing is also used to evaluate neuroblastic tumors, but it is not as specific as an MIBG study. [10, 27]
-
This frontal chest radiograph shows a right paraspinal well-defined density in the retrocardiac region. The right cardiophrenic angle appears normal.
-
T1-weighted magnetic resonance images of a ganglioneuroma. The sagittal views demonstrate a well-defined, solid mass located slightly anterior to the midthoracic vertebral bodies. The mass extends into and widening multiple neural foramina. Flow voids within the lesion represents vascularity.
-
T1-weighted postcontrast axial images of a ganglioneuroma demonstrate homogeneous enhancement of the lesion. The lesion is extending from the right paraspinal region into the epidural space through the neural foramina causing cord compression.
-
Frontal chest radiograph shows a large left paraspinal density. The lesion is silhouetting the aorta and, on the lateral view in the next image, appears to be retrocardiac and in close proximity to the vertebral column.
-
This lateral view radiograph of a ganglioneuroblastoma shows a large, well-defined homogeneous density that appears to be retrocardiac and in close proximity to the vertebral column.
-
This axial noncontrast computed tomography image of a ganglioneuroblastoma demonstrates a large, left heterogeneous paraspinal lesion with speckled calcifications that are predominantly peripheral. The mass is displacing the mediastinum to the right.
-
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
-
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina and is causing cord compression in the midthoracic region. The mass is also displacing the aorta to the right.
-
This coronal T2-weighted image demonstrates a well-defined, lobulated mass with fibrous septations. The mass is extending into the neural foramina.






