Practice Essentials
Ch'in and Tang first described cystic adenomatoid malformation (CAM) as a distinct entity in 1949. [1] Stocker in 2002 recommended the term congenital pulmonary airway malformation (CPAM) as being preferable to the term congenital cystic adenomatoid malformation, since the lesions are cystic in only 3 of the 5 types of these lesions and are adenomatoid in only 1 type (type 3). CCAM/CPAM is a developmental hamartomatous abnormality of the lung, with adenomatoid proliferation of cysts resembling bronchioles. CCAM/CPAM represents approximately 25% of all congenital lung lesions. [2, 3, 4, 5] The updated classification system of CPAM added types 0 and IV. Type 0 is very difficult to differentiate from bronchogenic cyst, The similarities between type IV and cystic pleuropulmonary blastoma may result in misdiagnosis. CPAMs are the most frequently seen congenital lung malformation, with an estimated incidence of 1 per 25,000 to 35,000 births. [6, 7]
The 5 Stocker classifications of CCAM (CPAM) are as follows [8, 9, 4, 10, 5] :
-
Type 0: the rarest form, 1-3% of cases, arises from the trachea or bronchus, presentation is severe and usually lethal, birth presentation, cysts of 0.5 cm with ciliated pseudostratified epithelial lining and goblet cells, bronchiolar cartilage present, all lobes involved.
-
Type I: 50–70% of cases, arising from the distal bronchus or proximal bronchiole, a small number of large echolucent cysts measuring 3–10 cm or a single dominant, cyst walls are thin and lined with a ciliated pseudostratified epithelium with bronchiolar differentiation, mucinous cells in 33% and cartilage in 10% of cases, only one lobe involved.
-
Type II: 15–30% of cases, arise from terminal bronchioles, first month of life presentation, multiple spaced cysts of 0.5-2.5 cm (sponge-like appearance) as well as solid area, ciliated cuboidal or columnar epithelial lining, absence of mucinous cells and cartilage, highest incidence of associated anomalies at up to 60%, involvement of usually only one lobe.
-
Type III: 5–10% of cases, in uterus or birth respiratory distress, bulky firm mass with adenomatoid appearance and cysts of 1.5 cm, ciliated cuboidal epithelial lining, absence of mucous cells and cartilage, involvement of usually only one lobe or one lung, associated with a poorer prognosis, microcystic.
-
Type IV: 5–15% of cases, newborn respiratory distress, pneumothorax, pneumonia or incidental finding, peripheral cysts with acinar-alveolar epithelial differentiation, cysts as large as 10 cm and associated with malignancy, specifically pleuropulmonary blastoma, alveolar in origin, antenatally classified as microcystic (< 5 mm).
(The following 2 radiographs are from the same infant with congenital cystic adenomatoid malformation.)
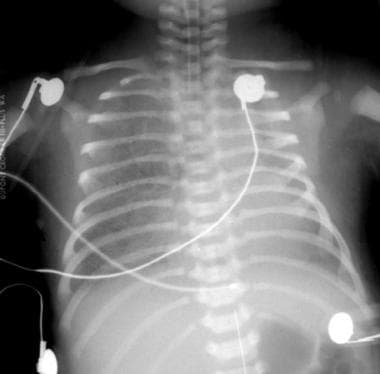 Initial anteroposterior radiograph of the chest in a patient with congenital cystic adenomatoid malformation on the first day of life, with dense lungs and a suggestion that the right lung is slightly more voluminous than the left lung.
Initial anteroposterior radiograph of the chest in a patient with congenital cystic adenomatoid malformation on the first day of life, with dense lungs and a suggestion that the right lung is slightly more voluminous than the left lung.
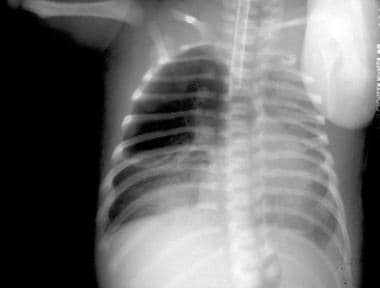 On the second day of life (same patient as in previous image), an anteroposterior radiograph shows physiologic fluid resorbed from an area of congenital cystic adenomatoid malformation and replaced with an air-containing cystic area occupying the right upper lung.
On the second day of life (same patient as in previous image), an anteroposterior radiograph shows physiologic fluid resorbed from an area of congenital cystic adenomatoid malformation and replaced with an air-containing cystic area occupying the right upper lung.
CCAM (CPAM) receives its blood supply from the pulmonary circulation and is not sequestered from the tracheobronchial tree. However, type II and III lesions can occasionally coexist with extralobar sequestration, and in such hybrid cases, they may receive systemic arterial supply. CCAM (CPAM) may also occur in combination with a polyalveolar lobe. A polyalveolar lobe is a form of congenital emphysema with increased number of alveoli with normal bronchi and pulmonary vasculature. CCAM (CPAM) usually occurs early in fetal life, whereas polyalveolar lobe occurs late. [11] Multiple lesions are uncommon and can be bilateral or multilobar in one lung.
The microscopic features that distinguish CCAM (CPAM) from normal lung include the proliferation of the terminal respiratory structures forming cysts; polypoid projections of the mucosa; increased smooth muscle and elastic tissue within cyst walls; the absence of cartilage; the presence of mucous-secreting cells; and the absence of inflammation. [5]
Differential diagnosis
CCAM (CPAM) is differentiated from other congenital cystic disease by 5 characteristics [12, 13, 14] :
-
Absence of bronchial cartilage (unless it is trapped within the lesion)
-
Absence of bronchial tubular glands
-
Presence of tall columnar mucinous epithelium
-
Overproduction of terminal bronchiolar structures without alveolar differentiation, except in the subpleural areas
-
Massive enlargement of the affected lobe that displaces other thoracic structures
Imaging modalities
CCAM (CPAM) may be initially detected during prenatal ultrasonography. [15, 8, 16] After birth, chest radiography should be performed first. [17] Although lesions remain filled with fluid, postnatal sonography can be used for a more detailed assessment, particularly in type III lesions. In a case series of 7 neonates with congenital pulmonary malformations, lung ultrasonography readily detected the lesions, and the findings were highly consistent with CT results. Once lesions are air-filled, CT scanning is necessary for determination of the type and extent of the lesions. [18, 19, 20, 21, 22]
Prenatally diagnosed lesions may be asymptomatic at birth (71%), and they have normal radiographic findings (57%). A concurrent sequestration may not be identified. Usually, radiographic findings are apparent in a symptomatic individual, but they may not be as apparent in an asymptomatic child. [23]
Most often, the diagnosis can be made by using plain radiographs. CT scans may be used to diagnose confusing cases. Overlapping CT features exist among cases of CCAM (CPAM), pulmonary sequestration, bronchogenic cyst, and other foregut malformations. CT is more accurate than radiography or ultrasonography in classifying the type of CCAM (CPAM). [3, 24]
In a retrospective study, Scialpi et al proposed that because of similar CT scan patterns between intrapulmonary bronchogenic cyst (IBC) and type I CCAM (CPAM) that precluded differentiation, surgical resection of all intrapulmonary cystic lesions in adults is required, as type I CCAM (CPAM) is a precursor of mucinous bronchioloalveolar carcinoma. [25] In the study, of 9 patients with a histologic diagnosis of pulmonary cystic disease following surgery, 6 had IBC and 3 had type I CCAM (CPAM).
Komori et al determined the optimal age for surgery in patients with congenital cystic lung disease is younger than 1 year. They used radionuclide imaging to assess long-term pulmonary function following lobectomy for congenital cystic lung disease in 93 infants and children. Patients who were younger than 1 year at surgery had a significantly lower mean transit time (marker for air trapping) at 5 years and 10 years postoperatively than those who were older than 1 year at surgery. Additionally, at 10 years after surgery, perfusion scintigraphy scores were higher and mean transit times were lower in patients without infection as compared to patients with infection. [26]
In a French study, Zeidan et al showed that prenatal MRI was less accurate than postnatal CT scan, which, according to the authors, remains the most reliable diagnostic modality to specify the location, extent, and type of lesions. [17] The authors evaluated the accuracy of prenatal MRI and postnatal CT imaging in identifying congenital cystic adenomatoid malformation and bronchopulmonary sequestration by comparison with histologic analysis.
Radiography
Usually, the radiographic pattern of CCAM (CPAM) appears as an expansile soft-tissue mass containing multiple air-filled cystic masses of varying size and shifting of the mediastinum. Initially and early in life, a homogeneous fluid-opacity pulmonary mass may present and evolve to demonstrate an air-filled cystic radiographic appearance. The initial dense appearance is a result of delayed emptying of alveolar fluid via either the bronchi or lymphatic and circulatory systems. Findings are usually apparent in a symptomatic individual, but they may not be as apparent in an asymptomatic child.
In patients with CCAM (CPAM), the pattern in the lung demonstrates multiple radiolucent areas that vary greatly in size and shape. Cysts are separated from each other by strands of opaque pulmonary tissue. The involved lung may appear honeycombed or spongy, but occasionally, one large cyst may overshadow the others.
Air-trapping within cystic spaces can cause rapid enlargement of the CCAM (CPAM) and subsequent respiratory embarrassment.
(See the radiographic images of cystic adenomatoid malformation, below.)
Initial anteroposterior radiograph of the chest in a patient with congenital cystic adenomatoid malformation on the first day of life, with dense lungs and a suggestion that the right lung is slightly more voluminous than the left lung.
On the second day of life (same patient as in previous image), an anteroposterior radiograph shows physiologic fluid resorbed from an area of congenital cystic adenomatoid malformation and replaced with an air-containing cystic area occupying the right upper lung.
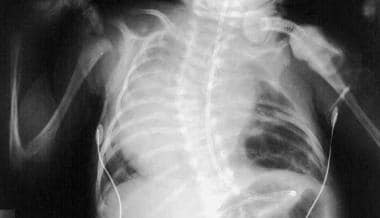 Anteroposterior radiograph of a patient with congenital cystic adenomatoid malformation with bilateral basilar cystic disease at several weeks of age. The patient's mediastinum is positioned toward the right, secondary to congenital scoliosis and larger left-sided cysts.
Anteroposterior radiograph of a patient with congenital cystic adenomatoid malformation with bilateral basilar cystic disease at several weeks of age. The patient's mediastinum is positioned toward the right, secondary to congenital scoliosis and larger left-sided cysts.
Most commonly, CCAM (CPAM) appears as a mass composed of numerous air-containing cysts scattered irregularly throughout a segment of the lung. The mass is space occupying, expanding the ipsilateral hemithorax and shifting the mediastinum to the contralateral side.
Usually, CT is not necessary for the diagnosis of CCAM (CPAM) in the neonatal period. Identifying the lobe involved or determining the extent of mass effect on the uninvolved lung may be possible. CT can be used to diagnose confusing cases encountered in infants, children, or adults or for planning surgery.
Pneumatoceles that form subsequent to bacterial pneumonia (eg, streptococcal, staphylococcal) can be mistaken for CCAM (CPAM), particularly in the older child.
Congenital lobar emphysema refers to overexpansion of 1 lobe, typically an upper lobe or right middle lobe, that leads to mass effect and respiratory distress. Although this entity could potentially be confused with CCAM (CPAM), typical features of overexpanded but normal parenchyma can be observed and confirmed with CT if necessary.
Pulmonary interstitial emphysema may resemble CCAM (CPAM) when it is complicated by large air collections. However, these are also typically associated with linear collections and preceded by high-pressure ventilation and barotrauma. The air collections are located in the interstitial lymphatics. On plain radiographs, intrapulmonary sequestration with infection and abscess formation can be difficult to differentiate from CCAM (CPAM).
Bronchogenic cysts are usually fluid filled and well circumscribed. Neuroenteric cysts are posterior mediastinal soft-tissue masses that are usually associated with vertebral anomalies.
Computed Tomography
CT findings of CCAM (CPAM) are correlated with the pathologic findings. [27, 28, 29] Areas of small cysts (< 2 cm in diameter) appearing with other abnormalities (a larger cystic area, consolidation, or low attenuation) are the most frequent findings.
Multiple large cystic lesions (>2 cm in diameter) are seen alone or with other abnormalities (areas of small cysts, consolidation, or low attenuation). Low-attenuation areas are clusters of microcysts. Air-fluid levels can be seen in some cysts. These lesions may be predominantly type I, type II, or a combination of both.
CCAM (CPAM) may completely resolve, as indicated by sonographic and plain radiographic criteria, but persistent abnormalities are well demonstrated on CT examination.
(See the CT images of cystic adenomatoid malformation below.)
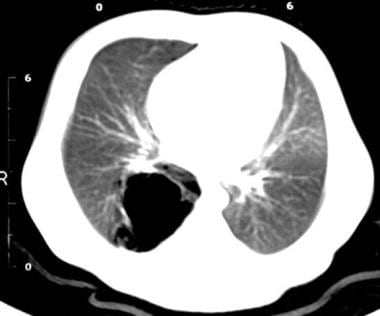 CT scan of the chest demonstrating a multiseptated cystic lesion in the right upper lobe consistent with localized congenital cystic adenomatoid malformation.
CT scan of the chest demonstrating a multiseptated cystic lesion in the right upper lobe consistent with localized congenital cystic adenomatoid malformation.
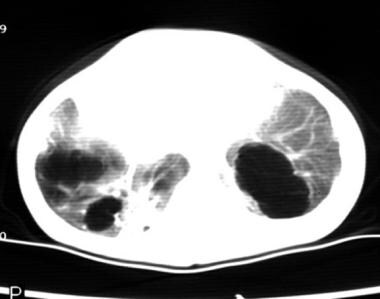 CT image of the bases of the lungs showing an unusual bilateral congenital cystic adenomatoid malformation.
CT image of the bases of the lungs showing an unusual bilateral congenital cystic adenomatoid malformation.
Approximately 25% of lesions diagnosed as CCAM (CPAM) may be either pulmonary sequestration or bronchogenic cysts. Overlapping CT features can also exist among other foregut malformations.
In a retrospecitive study of 33 CCAM (CPAM) patients, researchers found that patients with histologic signs of inflammation were more likely to have smaller tumors, whereas patients with mucinous proliferation (MP) were more likely to have larger tumors. In differentiating CCAM (CPAM) from mucinous adenocarcinoma in situ (AIS), the researchers found that while both lesions were associated with the presence of MIP, patients with AIS had significantly smaller lesions than patients with CCAM (CPAM). [30]
In a retrospective study, Scialpi et al proposed that because of similar CT scan patterns between intrapulmonary bronchogenic cyst (IBC) and type I CCAM (CPAM) that precluded differentiation, surgical resection of all intrapulmonary cystic lesions in adults is required, as type I CCAM (CPAM) is a precursor of mucinous bronchioloalveolar carcinoma. [25] In the study, of 9 patients with a histologic diagnosis of pulmonary cystic disease following surgery, 6 had IBC and 3 had type I CCAM (CPAM).
Zeidan et al showed that prenatal MRI was less accurate than postnatal CT scan, which, according to the authors, remains the most reliable diagnostic modality to specify the location, extent, and type of lesions. [17] The authors evaluated the accuracy of prenatal MRI and postnatal CT imaging in identifying congenital cystic adenomatoid malformation and bronchopulmonary sequestration by comparison with histologic analysis.
Magnetic Resonance Imaging
In CCAM (CPAM), prenatal MRI findings on T2-weighted images have been reported, [31, 32, 33, 34] such as the following:
-
CCAMs appear as intrapulmonary masses with increased signal intensity on T2-weighted images; type I or type II CCAM (CPAM) lesions have very high signal intensity almost equal to that of amniotic fluid and markedly higher than that of the surrounding unaffected lung tissue
-
With increasing numbers of microcysts or macrocysts, discrete cystic components may be seen within the mass lesion; cysts larger than 3 mm are visualized easily
-
Type III CCAM (CPAM) lesions have moderately high signal intensity; the signal intensity is higher than that of unaffected lung tissue but not as high as that of amniotic fluid; type III lesions are relatively homogeneous
-
MRI may be useful to fetal surgeons for planning the surgical approach when hydrops and polyhydramnios necessitate surgical intervention
Zeidan et al showed that prenatal MRI was less accurate than postnatal CT scan, which, according to the authors, remains the most reliable diagnostic modality to specify the location, extent, and type of lesions. [17] The authors evaluated the accuracy of prenatal MRI and postnatal CT imaging in identifying congenital cystic adenomatoid malformation and bronchopulmonary sequestration by comparison with histologic analysis.
Ultrasonography
Prenatal ultrasonography enables the identification of CCAM (CPAM) in a population of infants who are asymptomatic at birth. Regression of CCAM (CPAM) on prenatal sonograms is common, but this process usually does not continue postnatally.
Partially cystic, partially echogenic masses are characteristic of type I or type II lesions; the size or dimension of the cysts distinguishes the 2 types. Type III lesions may be large and entirely echogenic.
Usually, the newborn is symptomatic at birth, with the finding of a lesion exceeding 50% of the hemithorax.
The accuracy of prenatal ultrasonography in classifying the lesions of CCAM (CPAM) is approximately 77%. Prenatally diagnosed lesions may be asymptomatic at birth (71%), and there may be normal findings on radiographic examinations (57%). If radiographic results are normal, CT should be performed because it is more sensitive for detection of smaller lesions.
False-positive findings include bronchogenic cysts and pulmonary sequestration.
Nuclear Imaging
Komori et al used radionuclide imaging to assess long-term pulmonary function following lobectomy for congenital cystic lung disease in 93 infants and children, and they determined the optimal age for surgery in patients with congenital cystic lung disease is younger than 1 year. [26] Patients who were younger than 1 year at surgery had a significantly lower mean transit time (marker for air trapping) at 5 years and 10 years postoperatively than those who were older than 1 year at surgery. Additionally, at 10 years after surgery, perfusion scintigraphy scores were higher and mean transit times were lower in patients without infection compared with patients with infection.
-
Initial anteroposterior radiograph of the chest in a patient with congenital cystic adenomatoid malformation on the first day of life, with dense lungs and a suggestion that the right lung is slightly more voluminous than the left lung.
-
On the second day of life (same patient as in previous image), an anteroposterior radiograph shows physiologic fluid resorbed from an area of congenital cystic adenomatoid malformation and replaced with an air-containing cystic area occupying the right upper lung.
-
CT scan of the chest demonstrating a multiseptated cystic lesion in the right upper lobe consistent with localized congenital cystic adenomatoid malformation.
-
Anteroposterior radiograph of a patient with congenital cystic adenomatoid malformation with bilateral basilar cystic disease at several weeks of age. The patient's mediastinum is positioned toward the right, secondary to congenital scoliosis and larger left-sided cysts.
-
CT image of the bases of the lungs showing an unusual bilateral congenital cystic adenomatoid malformation.




