Practice Essentials
Craniosynostosis is the premature fusion of the cranial sutures. The condition can occur as an isolated defect or as part of a syndrome and is recognized in 2 forms: simple and compound. In simple craniosynostosis, only 1 cranial suture is involved; compound craniosynostosis involves 2 or more sutures. [1, 2] When sutures close prematurely, the structure of the skull becomes altered, resulting in an atypical shape of the skull and leading to increased intracranial pressure, respiratory and neurologic effects, and developmental issues. [3, 4, 5, 6] Imaging is required for the accurate diagnosis, surgical planning, posttreatment evaluation, and identification of coexisting anomalies and complications associated with craniosynostosis. [7, 8] (See the images below.)
Craniosynostosis occurs 4 times more frequently in males than in females. [9] The prevalence of craniosynostosis is 1 per 2000-2500 live births. Nonsyndromic craniosynostosis is present in 75% of cases, and syndromic in 25%. [10, 11] The craniosynostoses are classified on the basis of the affected suture, with the sagittal being affected in 55-60% of cases, coronal in 20-25%, metopic in approximately 15%, and lambdoid in 3-5%. [3, 4, 5, 6] There are approximately 180 identified syndromic forms, with the Muenke, Saethre-Chotzen, Pfeiffer, Crouzon, and Apert syndromes being the most common. [12]
Imaging modalities
Patients in whom craniosynostosis is suggested should undergo a careful clinical examination, with the clinician looking for abnormalities of the skull and extremities.
Plain radiography is the first radiologic step. Plain radiography quickly and simply identifies skull-shape abnormalities, which are seen in most patients with craniosynostosis. With this simple and inexpensive examination, usually all cranial sutures can be surveyed for patency.
The entire length of each suture is not always visible on plain radiographs, and some patients have only a small bony bar limiting growth at a particular suture. If the skull shape is entirely normal, craniosynostosis is unlikely.
Cranial ultrasound (CUS) is an alternative imaging modality. It offers excellent imaging of superficial structures, with the potential to confirm or exclude fusion of cranial sutures while avoiding exposure to ionizing radiation in the very young infant. The normal gap of a patent suture or the obliteration in craniosynostosis can be clearly demonstrated with CUS in children younger than 12 months. [13]
In a review by DeFreitas et al of 22 prenatal ultrasounds of infants known to have nonsyndromic craniosynostosis, the most common diagnoses were sagittal synostosis ( n = 11) and metopic synostosis ( n = 6). Prenatal ultrasound sensitivity was 82% and specificity 87%. [14]
Conventional cranial computed tomography (CT) scans with bone windows or 3-dimensional (3D) CT scans are frequently obtained to confirm bony abnormalities and to delineate any associated intracranial anomalies. Three-dimensional CT is the criterion standard for the evaluation of craniosynostosis. [15, 16, 17] CT scanning is considered to be expensive and may necessitate that the patient be sedated. [18]
Although 3D CT has superior diagnostic value, concerns remain about the hazards of radiation exposure in infants, who are are 2-10 times more radiosensitive than adults. To reduce radiation dose in children, several strategies and techniques are available, including iterative reconstruction techniques.
Three-dimensional (3D) craniofacial stereophotogrammetry has gained some popularity as a reliable, noninvasive, radiation-free imaging modality. It uses optical sensors to obtain multiple 2D images from different angles, and the 2D images are then reconstructed as a 3D digital model. [19]
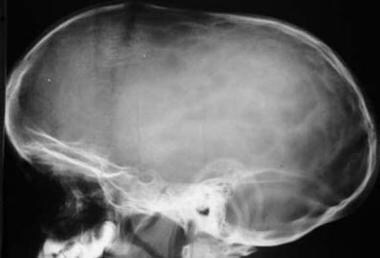 Sagittal synostosis. The anteroposterior (AP) diameter of the head is markedly increased (dolichocephaly), with flattening of the superior contour noted. The sagittal suture is fused, with widening of both the coronal suture and lambdoid suture.
Sagittal synostosis. The anteroposterior (AP) diameter of the head is markedly increased (dolichocephaly), with flattening of the superior contour noted. The sagittal suture is fused, with widening of both the coronal suture and lambdoid suture.
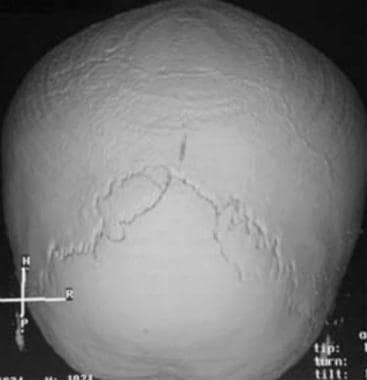 Three-dimensional CT scan demonstrates combined coronal and sagittal synostosis. Vertex view shows a normal lambdoid suture with complete fusion of the sagittal and coronal sutures.
Three-dimensional CT scan demonstrates combined coronal and sagittal synostosis. Vertex view shows a normal lambdoid suture with complete fusion of the sagittal and coronal sutures.
Guidelines
The Dutch Society for Plastic and Reconstructive Surgery published guidelines on craniosynostosis, including the following [20] :
-
Plain skull radiography is generally regarded as the first radiologic tool for diagnosis of craniosynostosis, but there is a clear role for ultrasound as a primary imaging modality for the detection or exclusion of craniosynostosis in children up to the age of 8 to 9 months. An ultrasound of the skull is considered the first radiologic diagnostic tool for children with skull-shape abnormalities who are suspected of having craniosynostosis (medium-risk craniosynostosis).
-
Low-dose 3D CT is considered the first radiologic diagnostic tool for children with high clinical suspicion of craniosynostosis (high-risk craniosynostosis).
-
A 3D CT scan is the most reliable imaging modality for diagnosing craniosynostosis, with higher diagnostic accuracy than ultrasound or radiography of the skull.
-
MRI is performed for syndromic craniosynostosis. Black bone MRI is a promising alternative to 3D CT scan of the skull in children with syndromic craniosynostosis, for whom an MRI examination to detect or exclude associated intracranial abnormalities is indicated. The disadvantage of black bone MRI is that the examination generally has to be performed under anesthesia.
-
For surgical planning, the standard use of a 3D CT scan of the skull to objectify the abnormality is highly recommended.
-
An additional MRI examination of the skull is of added value in children with syndromic craniosynostosis to detect or exclude associated defects of the brain and increased intracranial pressure, as well as assessment of aberrant venous vascular structures that may have implications for surgical planning.
Radiography
Plain radiographs are obtained easily and demonstrate osseous anatomy well. At a minimum, views should include anteroposterior (AP), Townes, and bilateral lateral films. Plain radiographs are useful for identifying the abnormalities of head shape (dolichocephaly, brachycephaly, and plagiocephaly) that are characteristic of the various forms of craniosynostosis.
(See the images below.)
Sagittal synostosis. The anteroposterior (AP) diameter of the head is markedly increased (dolichocephaly), with flattening of the superior contour noted. The sagittal suture is fused, with widening of both the coronal suture and lambdoid suture.
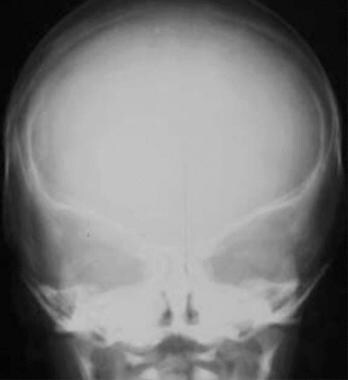 Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture. Note the bilateral harlequin configuration of the orbits (see also the slitlike appearance of the coronal suture in the next image).
Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture. Note the bilateral harlequin configuration of the orbits (see also the slitlike appearance of the coronal suture in the next image).
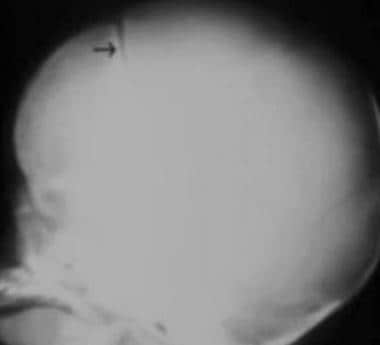 Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture (same patient as in the previous image). Note the bilateral harlequin configuration of the orbits and the slitlike appearance of the coronal suture (arrow). The margins of the coronal suture are densely sclerotic as well.
Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture (same patient as in the previous image). Note the bilateral harlequin configuration of the orbits and the slitlike appearance of the coronal suture (arrow). The margins of the coronal suture are densely sclerotic as well.
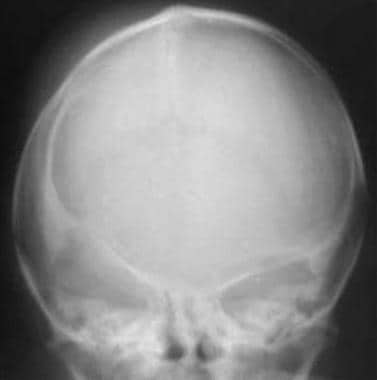 Combined synostosis also demonstrating plagiocephaly. AP view in a newborn with combined fusion of the sagittal and coronal sutures. Note the sclerotic margins and heaped-up bone of the fusing sagittal suture, the flattening of the right side of the calvaria (plagiocephaly), and the right harlequin orbit.
Combined synostosis also demonstrating plagiocephaly. AP view in a newborn with combined fusion of the sagittal and coronal sutures. Note the sclerotic margins and heaped-up bone of the fusing sagittal suture, the flattening of the right side of the calvaria (plagiocephaly), and the right harlequin orbit.
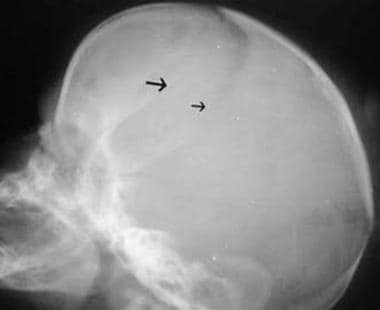 Combined synostosis also demonstrating plagiocephaly (same patient as in the previous image). Lateral view in a newborn with combined fusion of the sagittal and coronal sutures. The right coronal suture is abnormally straight (large arrow) and narrow in appearance, whereas the left suture is normal (small arrow).
Combined synostosis also demonstrating plagiocephaly (same patient as in the previous image). Lateral view in a newborn with combined fusion of the sagittal and coronal sutures. The right coronal suture is abnormally straight (large arrow) and narrow in appearance, whereas the left suture is normal (small arrow).
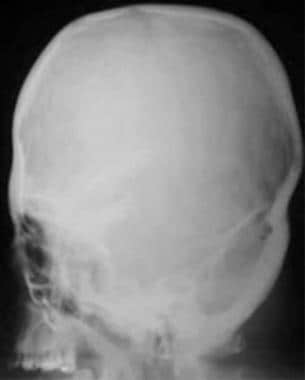 Apert syndrome. Markedly deformed tower-shaped head resulting from the premature fusion of all cranial sutures. Patient also had syndactyly, mitten hands, and sock feet.
Apert syndrome. Markedly deformed tower-shaped head resulting from the premature fusion of all cranial sutures. Patient also had syndactyly, mitten hands, and sock feet.
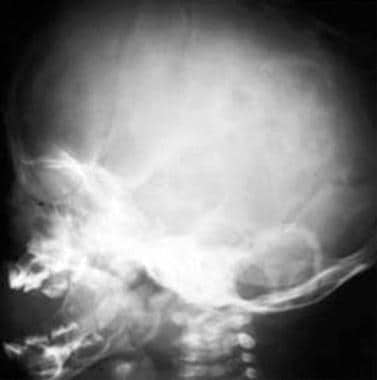 Crouzon disease. Note the abnormal shape of the head, with premature fusion of the sagittal suture and hypoplastic maxilla, which is severely disproportionate to the normal mandible.
Crouzon disease. Note the abnormal shape of the head, with premature fusion of the sagittal suture and hypoplastic maxilla, which is severely disproportionate to the normal mandible.
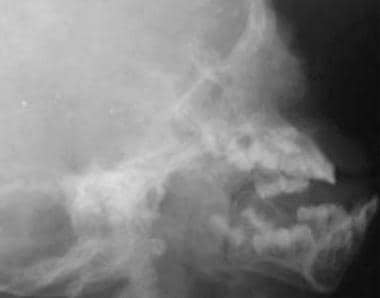 Crouzon disease. Patient had an abnormal shape of the head with premature fusion of the sagittal suture and hypoplastic maxilla (same patient as in the previous image). Clinically, the patient had severe proptosis due to underdeveloped orbits.
Crouzon disease. Patient had an abnormal shape of the head with premature fusion of the sagittal suture and hypoplastic maxilla (same patient as in the previous image). Clinically, the patient had severe proptosis due to underdeveloped orbits.
Plain radiographs can be used to identify prematurely fused sutures. Normal sutures are seen on plain images as serrated, nonlinear, lucent lines. Sutures in patients with craniosynostosis are usually straight with sclerotic heaped-up margins or are completely absent. The sclerotic margins may outline the sutures well and lead to the false impression that they are patent. Particular attention should be paid to the presence of this sclerotic margin and to focal sites of heaped-up margins, which are indicative of premature synostosis.
Plain radiographs can also be used to demonstrate overall morphology of the cranium and to identify the presence of localized problems (constricting bony bands restricting growth).
In addition, plain radiographs can be employed in identifying the presence of generalized problems (copper-beaten appearance, indicating elevated ICP). They can be used to identify other skeletal anomalies as well.
Visualizing the length of all sutures is not always possible, and suture closure may be difficult to detect unless it is accompanied by an abnormal head shape. Normal variations in the shape of the pediatric skull exist. For example, many formerly premature infants have long, narrow skulls resembling dolichocephaly but without sagittal synostosis. Many children also have asymmetrical flattening of the occiput caused by habitually lying on 1 side of the head, without underlying suture abnormalities; this is called positional molding. These 2 types of skull deformities are more common than craniosynostosis.
Computed Tomography
CT scans provide a more detailed method of visualizing intracranial pathology and detailed anatomy of the calvaria and brain parenchyma. In contrast to plain radiographs, the skull base is visualized well, and hard and soft tissues of the craniofacial skeleton can be studied in detail. The sensitivity of CT scans, when combined with physical examination and plain radiography, approaches 100%. Even on CT scans, the entire length of every suture may not be clearly visible, and normal variations in skull shape may pose a problem. [15, 16, 17, 21] After abnormal or suggestive plain radiographic findings are noted, CT scans with bone windows with or without 3D reconstruction are frequently requested prior to surgical therapy.
(See the images below.)
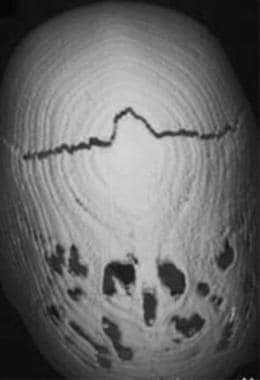 Three-dimensional computed tomography (CT) scan viewed from the top shows complete fusion of the sagittal suture, with a patent coronal suture and an elongated cranial contour. Apparent holes in the posterior parietal regions are due to normal thinning.
Three-dimensional computed tomography (CT) scan viewed from the top shows complete fusion of the sagittal suture, with a patent coronal suture and an elongated cranial contour. Apparent holes in the posterior parietal regions are due to normal thinning.
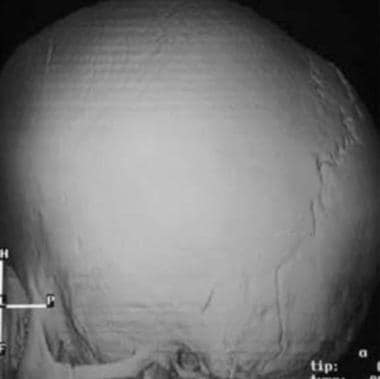 Three-dimensional CT scan shows brachycephaly. The AP diameter is shortened, with completely fused coronal sutures and open lambdoid sutures.
Three-dimensional CT scan shows brachycephaly. The AP diameter is shortened, with completely fused coronal sutures and open lambdoid sutures.
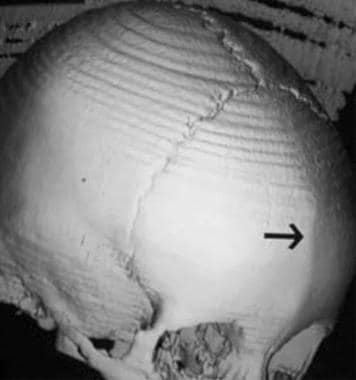 Trigonocephaly. Oblique view of the skull shows a ridge or keel in the midline of the frontal bone due to early fusion of the metopic suture (arrow).
Three-dimensional CT scan demonstrates combined coronal and sagittal synostosis. Vertex view shows a normal lambdoid suture with complete fusion of the sagittal and coronal sutures.
Trigonocephaly. Oblique view of the skull shows a ridge or keel in the midline of the frontal bone due to early fusion of the metopic suture (arrow).
Three-dimensional CT scan demonstrates combined coronal and sagittal synostosis. Vertex view shows a normal lambdoid suture with complete fusion of the sagittal and coronal sutures.
Neuroimaging is performed in children with isolated suture synostosis primarily to look for underlying brain damage or associated cerebral anomalies. Infants with trigonocephaly may have midline anomalies (eg, holoprosencephaly). Anomalies of the venous drainage and stenosis of the venous foramina at the skull base can occur with multisuture synostosis with syndrome- and nonsyndrome-related causes. Features such as shallow anterior fossa, deformed dystopic orbits, abnormal calvarial contour, and asymmetrical cranial base can be realistically depicted.
In a survey by Makar et al of 85 members of the American Society of Maxillofacial Surgeons and the American Society of Craniofacial Surgeons, the majority (63.5%) had performed a craniosynostosis operation in the past month, and only 9.4% never ordered preoperative CT scans. Of those who did order CT scans, the most common reasons were diagnosis confirmation (31.2%) and preoperative planning (27.3%). About 25% of surgeons ordered postoperative CTs—in most cases to evaluate surgical outcome(46.7%). [22]
Radiation dose
In a study comparing a lowered-dose CT protocol with a standard CT protocol in children evaluated for craniosynostosis, Ernst and colleagues found the effective dose of the low dose protocol was 0.08 mSv (97% reduction). Image quality was similar to the standard protocol in overall diagnostic acceptability, objective noise measurements, subjective cranial bone edge sharpness, and presence of artifacts. For objective sharpness of cranial bone-brain interface and subjective perception of noise, the images of the low-dose protocol were superior. [23]
In a phantom study, Kaasalainen et al reported sufficient image quality using a CT protocol with an effective radiation dose of 0.02 mSv, which is comparable to the effective dose of plain radiography (approximately 0.01 mSv to 0.04 mSv). [24] These results may be promising in children with complex craniofacial malformations who undergo repeated CT scans at the time of diagnosis and at various stages of surgical correction. [7]
Danelson et al investigated the possible benefits of employing 3D models in preoperative planning for craniosynostosis surgery—specifically, spring-mediated cranioplasty and cranial vault reconstruction. [21]
Magnetic Resonance Imaging
MRI shows better definition of intracranial soft-tissue structures than does CT scanning. In addition, MRI is useful in the detection of hydrocephalus and cerebral developmental defects, such as myelination defects and deformities of the maxilla resulting in airway compromise. [25, 12]
If children with craniosynostosis have abnormalities of tone or have diminished movements, MRI should be performed, because it is the most sensitive method for detecting cortical and white-matter abnormalities.
MRI is not a strong modality for evaluating bony abnormalities and thus cannot be used as the primary method of evaluating craniosynostosis. MRI is used primarily for assessing associated brainstem and soft-tissue abnormalities.
Leonhardt et al performed a prospective study on 20 patients with suspected craniosynostosis (mean age 1.26 ± 1.38 yr, 10 females) who underwent MR imaging using the T1SGRE ( 3D T1-weighted spoiled gradient echo-based) sequence, as well as 2 additiional patients who presented with skull fractures (0.5 and 6.3 yr, both male), and found substantial agreement between the diagnosis based on the T1SGRE and the final diagnosis. [26]
Black bone imaging
Black bone imaging, a gradient-echo MRI sequence with a low flip angle, has been identified as a promising radiation-free technique for imaging craniosynostosis in pediatric patients. The black bone sequence enhances the bone–soft tissue boundaries and can show normal patent cranial sutures as high signal structures, distinguished from the signal void of the cranial bones. The technique uses a low flip angle with short TR (repetition time) and TE (time to echo) to produce uniform contrast of the soft tissues, with densely black cortical bone. This technique was first described in by Eley et al. [27, 12, 28]
Ultrasonography
Data support cranial ultrasound (CUS) as an easy and feasible imaging technique for assessment of the cranial sutures. In a study by Linz et al, CUS confirmed a clinical diagnosis of craniosynostosis or plagiocephaly in a group of 411 infants. [29] CUS has been show to be able to differentiate fused or patent sutures effectively and to differentiate nonsynostotic pathology, such as positional skull deformities and molding, from craniosynostosis. Increase of the width of the cranial sutures on serial sonograms may provide evidence for a possible increase in ICP, which is a major complication of craniosynostosis. [7]
In a published series of 126 infants younger than 12 months assessed for craniosynostosis with cranial ultrasound, sensitivity was 100% and specificity 98% (95% confidence interval, 94-100%) when compared to 4-view radiography. [13]
A systematic review of 12 studies comprising 1062 ultrasound results that were used to diagnose craniosynostosis reported that specificity ranged from 86 to 100% and sensitivity ranged from 71 to 100%. [30]
Ultrasonography is user dependent, and therefore, inexperienced personnel can miss the diagnosis of craniosynostosis.
-
Sagittal synostosis. The anteroposterior (AP) diameter of the head is markedly increased (dolichocephaly), with flattening of the superior contour noted. The sagittal suture is fused, with widening of both the coronal suture and lambdoid suture.
-
Three-dimensional computed tomography (CT) scan viewed from the top shows complete fusion of the sagittal suture, with a patent coronal suture and an elongated cranial contour. Apparent holes in the posterior parietal regions are due to normal thinning.
-
Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture. Note the bilateral harlequin configuration of the orbits (see also the slitlike appearance of the coronal suture in the next image).
-
Coronal synostosis. The AP diameter of the head is shortened (brachycephaly), with partially fused coronal sutures and a widened sagittal suture (same patient as in the previous image). Note the bilateral harlequin configuration of the orbits and the slitlike appearance of the coronal suture (arrow). The margins of the coronal suture are densely sclerotic as well.
-
Three-dimensional CT scan shows brachycephaly. The AP diameter is shortened, with completely fused coronal sutures and open lambdoid sutures.
-
Trigonocephaly. Oblique view of the skull shows a ridge or keel in the midline of the frontal bone due to early fusion of the metopic suture (arrow).
-
Combined synostosis also demonstrating plagiocephaly. AP view in a newborn with combined fusion of the sagittal and coronal sutures. Note the sclerotic margins and heaped-up bone of the fusing sagittal suture, the flattening of the right side of the calvaria (plagiocephaly), and the right harlequin orbit.
-
Combined synostosis also demonstrating plagiocephaly (same patient as in the previous image). Lateral view in a newborn with combined fusion of the sagittal and coronal sutures. The right coronal suture is abnormally straight (large arrow) and narrow in appearance, whereas the left suture is normal (small arrow).
-
Three-dimensional CT scan demonstrates combined coronal and sagittal synostosis. Vertex view shows a normal lambdoid suture with complete fusion of the sagittal and coronal sutures.
-
Apert syndrome. Markedly deformed tower-shaped head resulting from the premature fusion of all cranial sutures. Patient also had syndactyly, mitten hands, and sock feet.
-
Abnormal soft-tissue and bony fusion of the toes in a patient with Apert syndrome.
-
Crouzon disease. Note the abnormal shape of the head, with premature fusion of the sagittal suture and hypoplastic maxilla, which is severely disproportionate to the normal mandible.
-
Crouzon disease. Patient had an abnormal shape of the head with premature fusion of the sagittal suture and hypoplastic maxilla (same patient as in the previous image). Clinically, the patient had severe proptosis due to underdeveloped orbits.



