Practice Essentials
Limb-girdle muscular dystrophy (LGMD) refers to a group of disorders that manifest as weakness and wasting of arm and leg muscles, with muscles of the shoulders, upper arms, pelvic area, and thighs being most frequently involved. [1] Genetic testing, creatine kinase (CK) studies, muscle biopsy, and histologic examination can be used in the evaluation of LGMD. Patients with muscular dystrophy should be managed through a clinic with access to specialties that address neuromuscular disorders, including physical therapy, occupational therapy, respiratory therapy, speech and swallowing therapy, cardiology, pulmonology, orthopedics, and genetics. [2]
The earliest descriptions of limb-girdle weakness are ascribed to Leyden [3] and Möbius [4] in 1876 and 1879, respectively. They described adult patients with a pelvic and femoral distribution of weakness and atrophy with a benign course. In 1884, Erb characterized a juvenile form of proximal muscle weakness. [5] Erb's patient had only shoulder-girdle weakness and atrophy, with sparing of other muscles of the body and a benign disease course compared with that described by Duchenne in the 1860s. Duchenne, a French physician, initially described a condition of progressive lethal wasting of degenerative skeletal muscle, which was later referred to as Duchenne muscular dystrophy. At that time, the differentiation between the spinal muscular atrophies and weakness associated with central nervous system disorders and primary muscle disease had not been established.
In 1891, Erb put forward the concept of muscular dystrophies as a primary degeneration of muscle and coined the term "dystrophia muscularis progressiva." [6] Erb's description was followed by various attempts at classifying these dystrophic disorders. In 1909, Batten classified primary muscle disease into the following 7 categories, which are still used today [7] :
-
Duchenne pseudohypertrophic variety
-
Erb juvenile weakness
-
Fascioscapulohumeral dystrophy [8]
-
Myotonic dystrophy
-
Mixed form
Between 1909 and 1954, many individual case reports of primary muscle disease with a limb-girdle distribution of weakness were published. In 1954, when Walton and Nattrass reported 105 cases of limb-girdle weakness associated with many other disorders, the nosologic entity of limb-girdle dystrophy was formally established. [11] Walton and Nattrass described the disease as a progressive muscle weakness with atrophy involving predominantly proximal muscles (eg, pelvis, shoulder). They described the disease as having a variable age of onset in the late first, second, third, fourth, or fifth decade of life; a slow clinical progression; and an autosomal recessive or autosomal dominant form of inheritance. See image below.
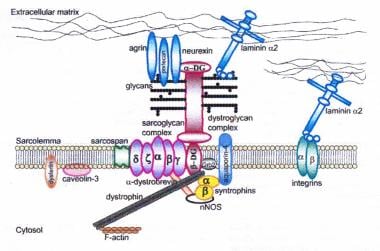 Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, in dysferlin, and in caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan, can result in limb-girdle muscular dystrophy.
Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, in dysferlin, and in caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan, can result in limb-girdle muscular dystrophy.
The development of sophisticated diagnostic tools of histology, histochemistry, ultrastructure, electrodiagnosis, and genetic studies has since shown that the entity, as originally described, is composed of a variety of neuromuscular disorders (eg, spinal muscular atrophy, polymyositis, endocrine disorders, metabolic conditions, congenital myopathies). Since the original descriptions of the condition, reports of many sporadic cases have been published with this pattern of muscle weakness associated with many other disorders.
Thus, the concept of LGMD as a nosologic entity was challenged, and now it is fair to consider it a symptom complex that consists of at least four disorders with varied inheritance patterns and etiologies. Therefore, importantly, the clinical features, the inheritance pattern, and the exclusion of other entities should define the disorders of LGMD.
Signs of limb-girdle muscular dystrophy
LGMD is suggested in patients who are toe-walkers and who have increased lumbar lordosis, forward pelvic tilt, and flexion and abduction of the hips. However, in the upper extremities, no typical features (eg, winging of the scapula) are present. Muscle hypertrophy is not a characteristic of the affected muscles.
Workup in limb-girdle muscular dystrophy
In patients with suspected muscular dystrophy, clinical phenotype—including muscle involvement pattern, inheritance pattern, age at onset, and associated disease manifestations—should guide genetic diagnosis.
The single biochemical abnormality in LGMD syndrome is CK level elevation. The CK elevation in the recessively inherited varieties of LGMD is significantly higher than in the rest of the LGMD spectrum.
Electromyographic abnormalities are atypical in LGMD, and electromyography (EMG) is more useful to exclude other disorders in the differential.
Muscle biopsy findings in LGMD are characterized by necrotic fibers with endomysial perivascular or perimysial mononuclear infiltration.
Hematoxylin and eosin and trichrome stains show a most striking predilection toward large fiber size in LGMD.
Management of limb-girdle muscular dystrophy
Given the slowly progressive nature of LGMD, the prudent approach to exercise therapy is to prescribe active-assistive and resistive movements and preserve and maintain muscle strength in the pelvic and shoulder girdle musculature. If the patient becomes nonambulatory, wheelchair mobility is essential.
Patients who develop an equinus foot deformity can benefit from tendon-lengthening surgery and/or knee-ankle-foot or ankle-foot orthoses to maintain mobility.
Pathophysiology
Molecular genetics of limb-girdle muscular dystrophy (LGMD)
Skeletal muscle consists of 2 major components: the sarcolemma and the sarcomeres. The sarcolemma is the sheath that covers the sarcomeres; it is composed of the plasma and basement membranes and the reticular lamina, which contains collagen. The sarcomeres represent the contractile element, which is composed of actin, myosin, and Z-band proteins (see image below). These proteins, like all others, are genetically coded and have a specific structure and distribution. Advances in molecular genetics have help in the discovery of significant information on the relationship between muscle biology and clinical neuromuscular diseases. This is very well exemplified in the shift from descriptive classifications of neuromuscular diseases to molecular pathobiologic classifications of neuromuscular diseases. [12, 13]
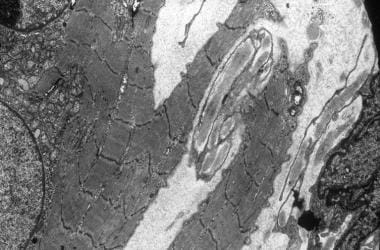 Electron micrograph showing streaming of band Z and splitting of the muscle fiber. A central nucleus is surrounded by a collection of small mitochondria.
Electron micrograph showing streaming of band Z and splitting of the muscle fiber. A central nucleus is surrounded by a collection of small mitochondria.
This concept is best observed in regard to our understanding of the very heterogeneous LGMD syndromes. These syndromes are now classified on the basis of at least 15 identified genes—5 autosomal dominant and 10 autosomal recessive. The 5 dominant genes are associated with the components of sarcomeres. The 10 recessive genes are associated with the plasma basement membrane and the adjacent reticular lamina, which contains the fibrillary collagen. [14, 15]
See the descriptions of each type of LGMD in the History section.
Epidemiology
Frequency
United States
Exact figures are not available. The frequency of limb-girdle muscular dystrophy in the general population cannot be estimated because of the heterogenous nature of this group of disorders (see Background).
Mortality/Morbidity
Limb-girdle muscular dystrophy is associated with low mortality and morbidity.
Race
No racial predilection is described for limb-girdle muscular dystrophy.
Sex
Limb-girdle muscular dystrophy may show an autosomal recessive or sporadic method of inheritance.
Age
Some forms of limb-girdle muscular dystrophy dramatically affect young adults, while other types progress so slowly that they are not detected until much later in life. [16]
-
Hematoxylin and eosin stain. Note the variation in fiber size. Necrotic fiber is shown with many nuclei (magnification 250X).
-
Marked endomysial fibrosis with atrophic and hypertrophic fibers.
-
Hematoxylin and eosin stain. Note the splitting of the fiber.
-
Gomori trichrome stain. Note the variation in fiber size and subsarcolemmal vacuoles, central nuclei, and subsarcolemmal collection of trichrome-positive material.
-
Light type I and dark type IIA fibers.
-
Electron micrograph showing abnormal mitochondria, a large lysosomal body, and a central nucleus.
-
Electron micrograph showing mitochondria with paracrystalline inclusions and lamellar bodies
-
Electron micrograph showing streaming of band Z and splitting of the muscle fiber. A central nucleus is surrounded by a collection of small mitochondria.
-
Trichrome stain. Note variation in fiber size. Necrotic fiber giant fibers and cytoplasmic inclusions.
-
Dystrophin-glycoprotein complex bridges the inner cytoskeleton (F-actin) and the basal lamina. Mutations in all sarcoglycans, in dysferlin, and in caveolin-3, as well as mutations that cause abnormal glycosylation of alpha-dystroglycan, can result in limb-girdle muscular dystrophy.








