Practice Essentials
Acid maltase deficiency (AMD) is an autosomal recessive disease characterized by an excessive accumulation of glycogen within lysosome-derived vacuoles in nearly all types of cells. Excessive quantities of free extralysosomal glycogen also have been described. AMD first was described by JC Pompe in Amsterdam in 1932; Pompe reported the case of a 7-month-old girl who became fatally ill from what appeared to be pneumonia. An autopsy revealed an unusually enlarged heart with normal valves. Pompe called this condition cardiomegalia glycogenica diffusa and considered it a disease analogous to von Gierke syndrome. The first article by Pompe was followed by similar reports by 2 independent authors, who described children with severe muscle weakness and cardiomegaly who died in early infancy. Their disease was attributed to an excessive deposition of glycogen in various tissues. This entity was named Pompe disease, and in 1957, GT Cori classified it as type II glycogenosis. [1] See the image below.
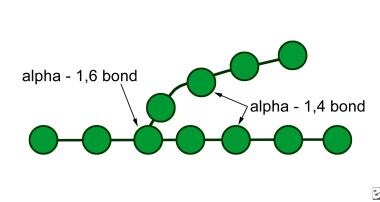 Glycogen molecule; by cleaving glycogen's 1,4 and 1,6 alpha-glycosidic linkages, the enzyme acid maltase gives rise to free glucose molecules.
Glycogen molecule; by cleaving glycogen's 1,4 and 1,6 alpha-glycosidic linkages, the enzyme acid maltase gives rise to free glucose molecules.
The following clinical phenotypes of AMD have been identified:
-
Infantile (Pompe disease)
-
Late infantile
-
Juvenile
-
Adult
Infantile acid maltase deficiency (Pompe disease) is the classic example of a metabolic myopathy and motor neuron disease that causes infantile hypotonia. This form of the disorder is the most severe and carries the worst prognosis, with death ensuing between ages 6 months and 2 years. The other forms are somewhat milder and vary in clinical presentation. [2, 3, 4]
Signs and symptoms of acid maltase deficiency
Infantile AMD is characterized by the following:
-
Early hypotonia
-
Massive cardiomegaly, soft murmur, and heart failure
-
Weakness and depressed or absent muscle stretch reflexes
-
Macroglossia
-
Moderate hepatomegaly
-
Intellectual disability
-
"Metabolic" anterior horn cell pathology (uncommon)
Juvenile AMD is characterized by the following [5, 6, 7, 8, 9] :
-
Motor delay
-
Progressive myopathy
-
Signs and symptoms limited to skeletal muscles
-
Respiratory insufficiency
Adult AMD is characterized by the following [5, 6, 7, 8, 9] :
-
Onset occurs in the second or third decade of life; muscle weakness progresses in the third to sixth decades of life
-
Proximal muscle weakness is greater than distal muscle weakness
-
The pelvic girdle is more involved than the shoulder; intercostal and diaphragmatic involvement is common
-
No liver, heart, or tongue enlargement occurs, except sometimes in terminal stages
Workup in acid maltase deficiency
Measurement of acid alpha-glucosidase enzyme activity in dried blood specimens is an optimal and reliable diagnostic test for acid maltase deficiency. [10] The tissue concentration level of acid maltase helps to establish a definite diagnosis. [11]
In patients with acid maltase deficiency, chest radiographs reveal an enlarged, globular heart associated with pulmonary vascular congestion. Atelectasis can also be seen.
With electromyography, myopathy is demonstrated, neuropathic findings may be revealed, and pseudomyotonia may be seen in the absence of myotonia.
Prenatal diagnosis using chorionic villi or amniocentesis is available for the fatal infantile form. Decreased serum levels of acid maltase can be detected. Acid phosphatase levels also can be measured and will be elevated.
Newborn screening for Pompe disease can be performed by determining the total acid alpha-glucosidase in plasma or dried blood spots.
With regard to biopsy, the diagnosis of AMD usually is made based on the absence or reduction in the levels of alpha–acid maltase in muscle tissue or cultured skin fibroblasts. This deficiency usually is more pronounced in the infantile form than in the juvenile and adult forms. [10]
Management of acid maltase deficiency
The use of assistive devices and orthoses may prove beneficial in patients with AMD who develop ambulatory difficulties. The use of intermittent positive pressure ventilation in Pompe disease would seem appropriate; however, because the disease involves not just the respiratory muscles but also the heart, the final outcome is likely to be the same.
Due to extended survival and musculoskeletal involvement, physiatric and orthopedic treatment may gain in importance in AMD.
Pathophysiology
Acid alpha-1,4 glucosidase (acid maltase), like other lysosomal enzymes, is synthesized as a precursor form (molecular weight 105,000) in the endoplasmic reticulum. The precursor molecule then is modified by the addition of a mannose-6-phosphate recognition signal that allows its transport to the lysosomes. Then, the acid maltase is partially degraded into a mature form with a molecular weight of 76,001. The gene for acid alpha-glucosidase is on chromosome band 17q23.
Acid maltase cleaves glycogen 1,4 and 1,6 alpha-glycosidic linkages. Its action gives rise to free glucose molecules (see History). [12, 13] See the images below.
Glycogen molecule; by cleaving glycogen's 1,4 and 1,6 alpha-glycosidic linkages, the enzyme acid maltase gives rise to free glucose molecules.
Nearly 35 distinct mutations have been identified in the q23-28 locus of chromosome 17, which encodes acid maltase. Establishing the genotype-phenotype correlation is difficult; however, the severity of mutation usually correlates with the severity of the disease. Deletions or missense mutations (mutations in which the base replacement changes the codon for one amino acid to that for another) usually are associated with the infantile variant (Pompe disease), whereas "leaky" (partial) mutations are associated with the childhood and adult forms of acid maltase deficiency.
The absence of acid maltase leads to an excessive accumulation of glycogen in lysosome-derived vacuoles. The presence of abnormal quantities of glycogen disrupts the normal architecture and function of the affected cells. The excess glycogen is expected to be, at least initially, in the vacuolar system. This has been found to be true in the liver and in other tissues; in muscle, however, most of the polysaccharide appears to be extravacuolar, possibly reflecting the fact that the glycogen is packed so densely in skeletal muscle that the surrounding membrane is difficult to see. Another possibility is that the intense pressure exerted on the vacuoles during muscular contracture causes them to rupture, allowing the contents to spill over into the cytosol.
Abnormal storage of glycogen occurs in many organs, including the central nervous system (CNS), heart, liver, and skeletal muscles, [2] thus leading to hypotonia; weak, bulky muscles; macroglossia; cardiomegaly; and congestive heart failure. The intramuscular storage of glycogen is more severe in Pompe disease than in any other glycogenosis.
Epidemiology
Frequency
United States
Pompe disease, or infantile acid maltase deficiency, occurs in 1 out of 50,000 live births.
Mortality/Morbidity
Pompe disease is inherited as an autosomal recessive disease. In the infantile form, death usually occurs between ages 6 months and 2 years; however, a less severe infantile form, with a better prognosis and improved survival, has been identified. Patients with the late infantile form may survive for several years. Patients with either the juvenile or adult form of acid maltase deficiency (each of which is also known as late-onset AMD) have been known to survive into the sixth or seventh decade of life. The clinical presentation may vary considerably, and some cases may go undetected; hence, the life expectancy for these groups is not exactly known.
Race
No ethnic predilection exists in connection with acid maltase deficiency.
Sex
Acid maltase deficiency occurs with equal frequency in males and females.
Age
The correlation of acid maltase deficiency with age depends on the form of the disease.
-
Glycogen molecule; by cleaving glycogen's 1,4 and 1,6 alpha-glycosidic linkages, the enzyme acid maltase gives rise to free glucose molecules.
-
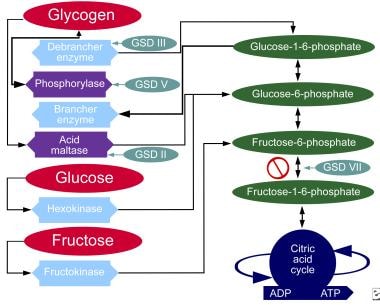
Metabolic pathways of carbohydrates.






