Overview
Most neuromuscular diseases (NMDs) are incurable. However, an effective rehabilitation program can help maintain a patient’s quality of life (QOL), as well as maximize the patient’s physical and psychosocial functions. An effective rehabilitation program can also minimize secondary medical comorbidity, prevent or limit physical deformity, and allow the patient to integrate into society.
Modalities such as range-of-motion and strengthening exercise, along with bracing and appropriate surgical intervention, may prolong ambulation. [1] Today, adaptive devices such as wheelchairs and lifts are now often interfaced with computer technology, providing better strategies for improving the patient’s mobility.
Endurance (aerobic) exercise may yield functional improvement and greater independence in activities of daily living (ADLs). Advancements in noninvasive positive-pressure ventilation technology have greatly reduced pulmonary morbidity in NMDs. Cardiac complications, though severe in some NMDs, often respond to medical management.
Psychosocial and vocational issues should be addressed as part of the management of NMDs. Such comprehensive management usually requires the efforts of a multidisciplinary team. This level of care is frequently provided in a tertiary care setting. This article discusses the key aspects of this type of care, which is critical to maximizing the quality of life for these individuals.
Definition and classification
An exact definition of what constitutes an NMD is important. Many clinicians who do not work directly in this field, even highly astute ones, incorrectly describe patients with NMDs as having muscular sclerosis (MS). In fact, MS is a demyelinating disease of the CNS; it is not an NMD, even though the symptoms of MS may sometimes superficially resemble the symptoms associated with NMDs.
NMDs are a group of diseases that affect any part of the nerve and muscle. These disorders include motor neuron diseases such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA), which may involve motor neurons in the brain, spinal cord, and periphery, ultimately weakening the muscle.
NMDs also include peripheral neuropathies such as Charcot-Marie-Tooth disease (CMT), which affect not only motor but also sensory nerves. Peripheral neuropathies start at the level of the dorsal root ganglion by definition. The neuromuscular junction may also be directly involved in diseases such as myasthenia gravis (MG).
Finally, NMDs may directly affect all forms of muscle, particularly skeletal and cardiac muscle. This stage of muscular deterioration is what causes the frequent misapplication of the term muscular dystrophy. The term dystrophy (from Greek dys-, “difficult or abnormal,” and trophe, “nutrition”) is also a misnomer based on descriptions from over 150 years ago, when lack of growth nutrients was blamed for damaging muscle. These disorders classified as muscular dystrophies fall under the broader category of myopathies (myo-, “muscle,” pathos, “disease”).
The term muscular dystrophy (MD) refers to a heterogeneous group of genetic disorders that typically result in progressive degeneration followed by incomplete regeneration of skeletal muscles, ultimately resulting in the loss of contractile tissue. In contrast to MD, the term myopathy generally refers to acquired or congenital muscle disorders that typically do not demonstrate ongoing cycles of degeneration/regeneration, but still result in weakness and disability due to loss of contractile function. Some MDs and myopathies affect cardiac and smooth muscle, in addition to other organs, including the brain.
The major forms of MD include Duchenne MD (DMD), Becker MD (BMD), limb-girdle MD (LGMD), facioscapulohumeral dystrophy (FSHD), myotonic dystrophy type 1 (DM1), myotonic dystrophy type 2 (DM2), congenital MD (CMD), oculopharyngeal MD (OPMD), distal MD (DMD), and Emery-Dreifuss MD (EDMD).
Some forms of MD appear at birth, while others become apparent during infancy, early childhood, adolescence, middle age, or later. The phenotypic severity is largely determined by the age at which the disorder first occurs, with early-onset disease typically resulting in more disability compared with late-onset disease. MD may result from spontaneous mutations, occurring de novo in an otherwise clear family line, or may be inherited. Forms of inheritance include autosomal dominant and recessive or sex linked.
Diagnosis and management
Accurate confirmation of the diagnosis is critical and involves thorough clinical evaluation, as well as electrodiagnostic studies and, often, muscle or nerve biopsy. For many of the diseases, DNA analysis of leukocytes or other cellular components obtained through a blood draw is commercially available and contributes greatly to the accuracy of the diagnosis.
Once the diagnosis has been confirmed, the patient and family members should be educated thoroughly about the expected outcome and the potential problems that may develop. The physician should then assess the patient’s and family’s goals and orchestrate a palliative and rehabilitative program that matches those goals. Enrollment in an experimental protocol should be encouraged and facilitated, furthering science and providing some hope for the patient.
Comprehensive management of this complex group of disorders is an arduous task under the best of circumstances. For this reason, a multidisciplinary approach, as mentioned above, is much more effective and takes advantage of the expertise of many clinicians rather than placing the burden on one. This team would consist of the following professionals, among others:
-
Physicians
-
Nurses
-
Physical, occupational, and speech therapists
-
Social workers
-
Vocational counselors
-
Psychologists
Treatment should be goal-oriented, with clear input regarding the patient’s expectations and personal goals. Although NMDs are not curable, they are treatable and do respond to rehabilitation. Ideally, because of the significant mobility problems associated with most NMDs, all key clinic personnel should be available at each visit. Tertiary care medical centers in larger urban areas can usually provide this type of service.
A number of organizations sponsor research and clinical care for people with NMDs, including the following (among others):
-
Muscular Dystrophy Association
-
Amyotrophic Lateral Sclerosis Association
-
Charcot-Marie-Tooth Association
-
Facioscapulohumeral Society
Governmental agencies that support research in NMDs include the National Institute on Disability and Rehabilitation Research, a division of the Department of Education, and the National Institutes of Health.
Clinical Characteristics of Neuromuscular Disease
This section provides with a brief clinical overview of the most common major neuromuscular diseases (NMDs). Any medical practice is likely to encounter these disorders. More in-depth discussions of the NMDs may be found in the articles specifically describing these disorders.
Motor neuron diseases
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is perhaps the most severe of all the major NMDs. It is a rapidly progressive NMD that destroys upper and lower motor neurons. This results in diffuse muscular weakness and atrophy. Unlike most primary nerve disorders, ALS also produces spasticity because of the loss of upper motor neurons. This creates unique clinical management issues.
An estimated 10% of all ALS cases are familial, usually inherited as an autosomal dominant trait. About 15% of these cases result from a gene defect on chromosome band 21q12.1, which leads to a mutation in the gene for the antioxidant enzyme Cu/Zn superoxide dismutase (SOD1). Approximately 100 SOD1 mutations have been identified, and nearly all are single missense dominant mutations causing toxic gain of function.
Over 50 unique superoxide dismutase mutations have been identified. Emerging evidence suggests that these mutations result in increased oxidative stress for the motor neurons, leading to cell death, which is presumably related to free radical toxicity. [2, 3]
However, most ALS cases occur sporadically, without a known cause. Studies suggest that an excess of the excitatory neurotransmitter glutamate in the central nervous system (CNS) is involved in the disease process. Serum, cerebrospinal fluid (CSF), and brain tissue of patients with ALS contain abnormally high levels of glutamate, apparently as a result of reduced clearance of glutamate from the motor cortex and decreased activity of glutamate transport proteins.
Population studies indicate that the prevalence of ALS is increasing, although this may be because of better recognition of the condition and increased longevity of people with ALS. [4] The worldwide prevalence is 5-7 cases per 100,000 population, making ALS one of the more common NMDs.
ALS seems to affect men more commonly than it affects women, with a male-to-female ratio of approximately 1.5:1. It primarily affects adults aged 40-60 years, with a mean onset age of 58 years. A higher prevalence of ALS exists in urban areas, possibly due to environmental factors. Considerable geographic clustering has been seen in association with ALS, most notably in the Western Pacific region of the world, but also in Gulf War veterans. Despite clustering, environmental or causal factors remain to be determined.
A population-based case-control study conducted in 3 counties of western Washington State showed that a history of smoking cigarettes was associated with a 2-fold increase in risk, whereas a 3-fold increase was seen in current smokers. The duration (years having smoked) and amount of smoking (packs per year) correlated with risk for ALS.
Dietary fat increased the risk for ALS, although alcohol consumption did not. Dietary fiber intake decreased risk, although consumption of antioxidant vitamins from diet or supplement sources did not alter the risk. Notably, glutamate consumption did correlate with an increased risk for ALS. That smoking and glutamate consumption are risk factors for ALS supports the theory implicating oxidative stress and excitotoxicity in the pathogenesis of ALS.
The major neuropathologic finding in ALS is the degeneration and subsequent loss of motor neurons as a consequence of apoptosis (programmed cell death). Apoptosis is characterized by neuronal contraction (to approximately one fifth the normal size) with an extremely condensed nucleus and cytoplasm. Apoptotic bodies can usually be seen in macrophages.
Other neuropathologic findings in ALS include axonal loss in the descending motor tracts, anterior roots, and nerves. There also appears to be subtle involvement of the frontal lobes, hippocampal area, substantia nigra, and dorsal columns.
Motor neuron degeneration begins focally and spreads to contiguous regions in the neuraxis until the neurons controlling respiration are affected, which ultimately leads to death from respiratory failure. The number of motor neurons involved and the spectrum of motor neuron degeneration varies for any individual patient, which accounts for the clinical variability in disease progression.
Several prognostic predictors exist for determining the course of ALS. Presentation with bulbar or pulmonary dysfunction, a short interval from symptom onset to diagnosis, electrodiagnostic findings primarily involving lower motor neurons, and advanced age all potentially indicate a poor prognosis. Women present with bulbar symptoms more often than men do. Bulbar palsy appears to progress more rapidly in women; this indicates a poor prognosis. Young males with ALS have the best prognosis and may have a longer life expectancy.
Overall, the median 50% survival rate is 2.5 years after diagnosis. In patients who present with bulbar symptoms, the 50% survival rate drops to 1 year. Survival rates vary, depending on the patient’s decision to use a feeding tube and assisted ventilation. Nonetheless, by 5 years after diagnosis, the overall survival rate is only 28%.
A review by Benatar et al of randomized, controlled trials sought to determine whether treatment response differed between patients with familial ALS and persons with the sporadic form of the disease. [5] Analyzing data from 4 trials that collectively included 41 patients with familial ALS and 822 patients with the sporadic condition, the authors found no statistically significant difference in treatment response between the 2 groups.
Spinal muscular atrophy
All forms of spinal muscular atrophy (SMA) involve selective destruction of anterior horn cells. The distinct types of SMA differ clinically. Some rare forms affect only distal or bulbar muscles. SMA is usually classified as types I, II, and III. Most forms of SMA are autosomal recessive traits.
SMA I, also known as Werdnig-Hoffmann disease or acute infantile-onset SMA, is a severe disorder that causes death before age 2 years (see the image below). SMA II, also known as chronic Werdnig-Hoffmann disease or early-onset intermediate SMA, is less severe. SMA II may not become apparent until age 6-18 months.
 Werdnig-Hoffman disease (type of spinal muscular atrophy). Small muscle fibers within separate muscle fascicles.
Werdnig-Hoffman disease (type of spinal muscular atrophy). Small muscle fibers within separate muscle fascicles.
SMA III, also known as Kugelberg-Welander disease (see the image below), has a much later onset (typically, age 5-15 years) and is associated with much less morbidity. [6, 7]
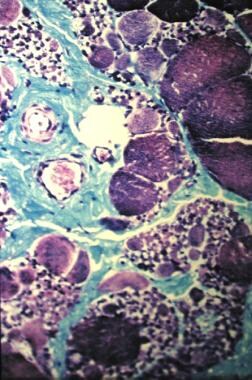 Kugelberg-Welander disease (type of spinal muscular atrophy). Marked variation in muscle fiber size, along with increased perimysial connective tissue.
Kugelberg-Welander disease (type of spinal muscular atrophy). Marked variation in muscle fiber size, along with increased perimysial connective tissue.
Mutations in exons 7 and 8 of the telomeric survival motor neuron gene are present in more than 98% of patients with SMA types I-III. Deletions in the neuronal apoptosis inhibitory protein gene are found in about 67% of patients with SMA I, 42% of patients with SMA II or III, and some patients with adult-onset SMA, though the precise percentage is not known.
Commercial blood tests (DNA analyses) are now available for use in diagnosing SMA. Prevalences for SMA types II and III have been estimated to be as high as 40 cases per million in the general population, though considerable variations exist in demographic studies. [4]
Two forms of later adult-onset SMA exist. The first type is spinobulbar muscular atrophy (SBMA), or Kennedy disease. This disorder, which was first described as recently as 1968, is a sex-linked recessive NMD characterized by progressive spinal and bulbar muscular atrophy, gynecomastia, and reduced fertility.
SBMA has been mapped to the androgen receptor on the X chromosome. The mutation, which consists of an expansion of cytosine-adenine-guanine (CAG) trinucleotide repeats, occurs in the first exon of the gene, producing decreased sensitivity of androgen receptors on motor neurons. The disease has some clinical variability; however, phenotypic expression does not correlate with the length of CAG repeats.
In this way, SBMA differs markedly from myotonic muscular dystrophy and fragile X syndrome, in which an increased number of tandem triplet repeats correlates directly with disease severity. SBMA can occur without any family history or gynecomastia, and all males with atypical ALS should undergo DNA testing for SBMA (the DNA test is commercially available).
The other form of later adult-onset SMA has its onset in patients aged 17-55 years, with either recessive or dominant types of inheritance. This form of SMA clinically resembles SMA III but may be more progressive. It has been mapped to chromosome band 5q11.2-13.3; however, commercial testing is not yet available, because adult-onset SMA and SBMA are far less common forms of SMA.
Peripheral neuropathies
Charcot-Marie-Tooth disease
Charcot-Marie-Tooth disease (CMT) can be divided into 2 basic types: primarily demyelinating (with secondary axonal loss) and primarily axonal. The remainder of the subclassification of CMT is based on genetic analysis.
In CMT type 1 (CMT1), which is primarily a demyelinating neuropathy, anatomic changes directly affect the myelin sheath, with secondary axonal changes. In areas of focal demyelination, impulse conduction from one node of Ranvier to the next is slowed; current leakage occurs and the time for impulses to reach threshold at successive nodes of Ranvier is prolonged, producing slowing of conduction velocity along the nerve segment.
CMT type 2 (CMT2) is a primary axonal neuropathy producing changes in the axon and the nerve cell body. Clinically, CMT2 is often less severe than CMT1. Patients with CMT2 may have more lower extremity involvement, although in other respects they may not be easily distinguishable from patients with CMT1. Most of the phenotypic descriptive studies in CMT were done before the advent of DNA testing.
Overall, CMT is a slowly progressive disorder characterized by diffuse muscle weakness and prominent distal atrophy, predominantly involving the intrinsic muscles of the feet and the peroneal muscles. Subjects with CMT produce 20-40% less force than normal controls on quantitative isometric and isokinetic strength measures, even though manual muscle test scores may be normal. No significant side-to-side difference exists with regard to strength. From a functional standpoint, the sensory deficit is usually less severe than the motor deficit.
Prior studies have also documented that subjects with CMT have a marked reduction in functional aerobic capacity during exercise testing, despite having normal or relatively normal preexercise pulmonary function, exercise heart rate, and blood pressure and maximum ventilation.
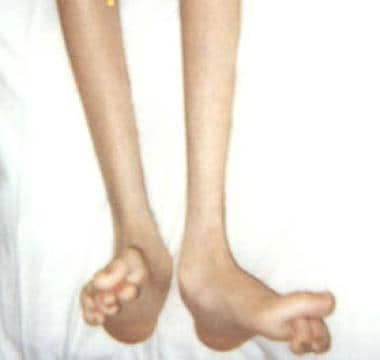
The number of molecular forms of CMT and related neuropathies is always growing. However, CMT1 is the most common type overall. CMT1A (see the image below) is the most common subtype of CMT1 and results from a duplication of chromosome segment 17p11.2, which contains the gene for peripheral myelin protein 22 (PMP22). [8]
Notably, patients with a related disorder, hereditary neuropathy with liability to pressure palsies (HNPP), show a large deletion, rather than a duplication, in the PMP22 gene. [9] HNPP is an autosomal dominant disorder that produces episodic recurrent nerve compression with focal demyelination at common sites of compression or entrapment (eg, wrist, elbow, and fibular head). Nerve compression can occur in the absence of true entrapment.
CMT X is an X-linked dominant, primarily demyelinating neuropathy with a mutation in the connexin 32 gene (CX32), which codes for a membrane protein (gap junction protein, beta 1) involved in the formation of gap junctions. CMT X1 is clearly a distinct entity. Some varieties of CMT X1 may exhibit abnormal temporal dispersion and heterogeneous conduction velocities that are very atypical of other hereditary neuropathies.
Mutations in the CX32 gene can produce a neuropathy with either demyelinating or axonal electrodiagnostic features. Some clinical and electrodiagnostic data in males with different missense mutations in the CX32 gene appear to differ significantly. [10] Furthermore, males with nonsense mutations have an earlier onset and a more severe phenotype than males with missense mutations.
Point mutations in PMP22 or the myelin protein zero gene (MPZ) may cause Dejerine-Sottas disease. [11] Thus, many cases of Dejerine-Sottas disease are now considered severe phenotypes within the genotypic spectrum of CMT1. Congenital hypomyelinating neuropathy is a severe and often fatal newborn disorder that presents with respiratory distress at birth and has been linked to the early growth response gene 2 (EGR2) in some families.
Muscular dystrophies
Duchenne MD and Becker MD (DMD/BMD)
The most common MD in children is DMD, an X-linked recessive disorder affecting approximately 1 in 3500 boys. DMD is caused by mutations in the gene coding for the sarcolemmal-spanning protein, dystrophin. In the absence of dystrophin, the muscle membrane becomes fragile, unable to maintain selective permeability, and ultimately leading to progressive necrosis and replacement with fat and connective tissue. [12, 13]
BMD is also caused by mutations in dystrophin, the same gene as DMD. However, in BMD, there is production of some dystrophin, although it may be a truncated protein. Compared with DMD, BMD causes a less severe phenotype. Most boys with DMD are wheelchair users by age 12 years and are profoundly disabled with a markedly reduced lifespan. Boys with BMD may ambulate into adulthood, although most develop cardiomyopathy. Both DMD and BMD are referred to as dystrophinopathies.
Limb-girdle MD (LGMD)
Sharing characteristic clinical features, LGMDs include a predilection for certain ethnicities; type of inheritance (autosomal dominant, recessive, or X-linked); patterns of weakness (limb girdle, humeroperoneal, or distal); hypertrophy or atrophy of specific muscle groups; and ancillary characteristics, such as scapular winging, level of serum creatine kinase (CK), cardiac and respiratory involvement, and characteristic histopathological features on muscle biopsy. Most LGMDs are caused by mutations in genes encoding dystrophin-associated sarcoglycans and may be referred to as dystroglycanopathies. Currently, more than 20 subtypes of LGMD are recognized, including autosomal dominant forms (LGMD type 1, A to H) and autosomal recessive forms (LGMD type 2, A to N), with some forms showing abnormalities on brain imaging. [14]
Facioscapulohumeral MD (FSHD)
FSHD is caused by a mutation in a yet-identified gene located near the end of the long arm of chromosome 4, referred to as 4q35 in the D4Z4 DNA region. FSHD has a high spontaneous mutation rate with up to one third of cases occurring outside a known family line. Moreover, about 2-5% of FSHD cases appear not to be linked to the 4q35 deletion on chromosome 4.
FSHD produces selective weakening of specific skeletal muscles, including those reflected in its name: face, shoulder girdle, and upper arms. Early loss of the ability to pucker the mouth and whistle or open and close the eyes, along with axial musculoskeletal deformities that often are painful, are some of the more characteristic features of FSHD. Progression is quite variable and usually gradual, and it may be documented on MRIs of affected muscles. [15] In more progressive forms, other skeletal muscles weaken, including pelvic, hip girdle, and abdominal muscles, and FSHD may be completely disabling. Approximately half of those with FSHD are able to maintain ambulation throughout their lives, and most live a normal life span.
Myotonic MD (or Steinert disease)
Myotonic MD (or Steinert disease) is the most common form of adult-onset MD, and it has 2 known forms, myotonic dystropy type 1 (DM1) and myotonic dystropy type 2 (DM2). [16] Both forms are autosomal dominant, affecting both men and women. DM1 is caused by mutations in the DMPK gene, while DM2 results from mutations in the CNBP gene. While the exact functions of these genes are not clear, the protein product of the DMPK gene appears to play a role in intracellular communication within skeletal muscle cells and in cells in the heart and brain. Structural changes in the DMPK and CNBP genes cause a segment of DNA to be abnormally repeated many times, resulting in instability in the gene. [17, 18, 19, 20] This leads to an abnormally expanded messenger RNA, which ultimately formstoxic intracellular clumps that interfere with the production of other critical proteins.
Rarely DM1 appears in newborns, producing a severely disabling disorder known as congenital DM (C-DM). In addition to weakness, a predominant symptom is myotonia, a delayed relaxation of muscles following contraction, which patients may describe as spasm or stiffness. Age of onset and severity of symptoms are dependent on the number of abnormal DNA repeats. DM1 may appear at birth (C-DM) up to early adulthood. [18] Very mild cases may escape clinical detection but may be diagnosed by DNA analysis or needle electromyography (EMG), which reveals characteristic myotonic “dive bomber” discharges. DM1 may also show a false-positive decrement on repetitive stimulation, yet may also rarely be associated with myasthenia gravis. DM1 may also be associated with diffuse musculoskeletal pain.
DM2 is caused by a mutation in the CNBP (ZNF9) gene. CNBP intron 1 contains a complex repeat motif, (TG)n(TCTG)n(CCTG)n. Expansion of the CCTG repeat causes DM2. DM2 is a milder disease, with later onset. DM2 produces muscle weakness, but also affects the CNS, heart, gastrointestinal tract, eyes, and endocrine glands. Reports have described genetically confirmed DM2 that has not revealed myotonia on EMG.
Congenital MD (CMD)
CMD is a heterogeneous group of disorders with a dystrophic pattern on muscle biopsy. CMD is present at birth or during the first months of life. The most common forms of CMD are caused by a laminin α-2 (merosin) deficiency. [21] Other forms are Ullrich congenital muscular dystrophy, fukutin-related proteinopathy, rigid spine syndrome, and glycosylation disorders of α-dystroglycan. The latter group is often associated with CNS defects, including lissencephaly, pachygyria, cerebellar and brainstem abnormalities, and variable ocular anomalies. [22]
Oculopharyngeal MD (OPMD)
OPMD is caused by a mutation in the PABPN1 gene [(GCN)(n)/polyalanine]. OPMD is rare and is associated with filamentous intranuclear inclusions (INIs), which may cause aggregation of mRNA, although it is not clear if this plays a pathologic role.
OPMD is a late-onset (40 y and older), slowly progressive disorder producing weakness in the eye and facial muscles, often leading to visual impairment and clinically significant dysphagia. Choking and recurrent aspiration pneumonia may occur, requiring gastrostomy tube placement. Weakness in pelvic and shoulder muscles may occur later.
Emery-Dreifuss MD (EDMD)
(EDMD has both X-linked and autosomal inherited forms. The X-linked form is caused by mutations in the gene encoding emerin, leading to deficiencies in nuclear envelope proteins. Autosomal forms are due to mutations in genes encoding for lamins A and C and are usually dominant.
EDMD is characterized by progressive muscular weakness, joint contractures, and cardiac disease, possibly life threatening. Elbow contractures occur early in EDMD.
Distal MD
This group of rare diseases affects adult men and women. Miyoshi-type distal muscular dystrophy (MDMD) is one of the more common forms, occurring in early adulthood. [23] MDMD is a dysferlinopathy caused by a mutation in the dysferlin gene (DYSF), very similar to LGMD type 2B.
MDMD and other forms of distal MD typically cause slowly progressive weakness in distal muscles and are usually less severe forms of MD.
Management of Neuromuscular Disease
Respiratory dysfunction
Restrictive lung disease, despite its name, does not affect the lung. People with restrictive lung disease develop “weak bellows”—that is, their breathing muscles (diaphragm, chest wall, and abdominal muscles) are weak. Thus, patients with neuromuscular disease (NMD) may have difficulty inhaling and exhaling; this also includes coughing. This is a common problem in many NMDs but is typically most severe in amyotrophic lateral sclerosis (ALS), Duchenne muscular dystrophy (DMD), spinal muscular atrophy (SMA), and myotonic muscular dystrophy (MMD).
Respiratory failure in fascioscapulohumeral dystrophy (FSHD) is not typically seen. However, a study identified 10 patients with FSHD on nocturnal ventilatory support at home, representing approximately 1% of Dutch people with FSHD. Severe muscle disease, wheelchair dependency, and kyphoscoliosis appeared to be risk factors for respiratory failure in FSHD.
There are numerous reported cases of respiratory failure in people with Charcot-Marie-Tooth disease (CMT), the etiology of which has remained elusive. Electrodiagnostic and pathologic studies on the phrenic nerve in patients with CMT confirm that respiratory failure is involved in the disease. [24]
In one study, phrenic nerve latency was abnormally prolonged in 96% of the subjects with CMT, but significant abnormalities in the results of pulmonary function tests and clinical symptoms were uncommon and did not correlate with the phrenic nerve latencies. [24] Although phrenic nerve latencies are markedly prolonged in CMT, they are not useful in predicting respiratory dysfunction.
The results of routine pulmonary function tests, including forced vital capacity (FVC) and maximal inspiratory and expiratory pressure, should be monitored closely. Maximal inspiratory pressure reflects diaphragm strength and ventilatory ability. Maximum expiratory pressure is indicative of abdominal and chest wall muscle strength and the ability to cough and clear secretions. Peak cough flow is another simple measurement that indicates the amount of pressure a patient can generate during a volitional cough.
The biggest problem in NMD is hypoventilation, which leads to hypercapnia (elevated carbon dioxide levels in the blood). End-tidal carbon dioxide levels or arterial blood gases should be periodically measured, depending on the clinical circumstances. Pulse oximetry, which measures only oxygen saturation levels, may be inadequate.
Performing a thorough review of systems is important. Patients who are hypoventilating become hypercapnic at night and experience a morning headache as a result. They may also have nocturnal restlessness, nightmares, and poor overall quality sleep, which result in daytime somnolence. Insufficient respiration with hypoxia may also develop; this usually occurs much later in the disease process, particularly in cases of lung damage involving chronic pneumonias, infections, or aspiration.
Patients should be educated early in the disease process so that informed decisions can be made further down the line. [25] Many options are available for noninvasive intermittent positive-pressure ventilation (NIPPV). Bimodal positive airway pressure (BiPAP) is generally considered the preferred modality of assisted ventilation in NMDs. BiPAP is similar to the older technology of continuous positive airway pressure (CPAP), which is used to treat sleep apnea.
In CPAP, inhalation and exhalation are assisted with continuous positive airway pressure (which feels like breathing into a stiff headwind). BiPAP cycles the pressure down on exhalation, though a net positive pressure gradient remains (as if the wind died down a bit, but not completely, during exhalation). Typical pressures would be 8-10 cm H2 O for inhalation and 4-6 cm H2 O for exhalation.
BiPAP can easily be used in the home, but some work with a respiratory therapist may be required to achieve a good face or lip seal on the pilot-type mask or nasal-oral orthotic interface that is typically used. If a mask fails to fit well or is too uncomfortable, a nose plug interface may be used, although this may not work as effectively.
Patients who use assisted oral ventilation, mainly at night, may initially avoid the need for tracheostomy and maintain a reasonable quality of life (QOL). However, bulbar palsy may occur in ALS and in some rare forms of SMA. In such cases, if better airway access becomes necessary and an informed patient wishes more aggressive care, a tracheostomy may be performed.
However, an alternative procedure, laryngeal diversion (or laryngotracheal separation), has several distinct advantages over tracheostomy. In laryngeal diversion, the trachea is surgically separated, and a cutaneous tracheostoma is formed with the distal segment. The proximal trachea is sewn either over or side-on-end into the esophagus. This completely eliminates the possibility of aspiration and requires much less deep suctioning than a tracheostomy does. [26]
The tracheostoma does not require any hardware (eg, a tracheostomy tube), and the patient may still take some food for pleasure without risking aspiration. The primary disadvantage is the complete loss of phonation, given that air no longer flows through the vocal cords. Accordingly, this procedure is recommended only when severe dysarthria accompanies the dysphagia and the patient’s speech is unintelligible.
Although a tracheostomy preserves the ability to phonate, it actually increases the risk of aspiration, requires significantly more care, and provides no better airway access. Both tracheostomy and laryngeal diversion facilitate the use of mechanical ventilation, but the patient must understand that neither procedure guarantees a better QOL. Fortunately, with the advances in noninvasive ventilation, these surgical options are now infrequently used.
In addition to assisted ventilation, various newer technologies may be used to improve respiratory hygiene. These include cough assist machines that help NMD patients bring up secretions; the machines produce an artificial cough via a face mask by rapidly changing airway pressure from positive to negative. Several of these products are commercially available in the United States, including the Cofflator (Respironics, Pittsburgh, PA) and the In-Exsufflator (JH Emerson Co, Cambridge, MA).
Dysphagia and dysarthria
Dysphagia and dysarthria may occur in ALS and some rare forms of SMA because of the involvement of the bulbar musculature.
Early signs of dysphagia include a hoarse voice and persistent cough, particularly after liquids are swallowed; this may indicate microaspiration. At the first sign of dysphagia, consult a speech language pathologist, who can perform clinical swallowing evaluations and make recommendations on dietary modification and safe swallowing strategies, such as the following [27] :
-
Thickening liquids
-
Using techniques such as double swallows, chin tucks, and head turns
-
Eating only foods that easily form into boluses
A modified barium swallow study, in which the patient swallows various textures of solid food and liquid laced with barium, is helpful for accurately determining the presence of aspiration, as well as for defining which food textures the patient can safely swallow. However, this study does involve exposure to radiation. An alternative to the modified barium swallow is flexible endoscopic evaluation of swallowing (FEES), a test that uses an endoscope specifically designed to assess the swallowing mechanism.
FEES directly evaluates motor and sensory components of swallowing through direct visualization of the reaction of the larynx to a stimulus delivered by the endoscopic camera, which can then photograph the reaction. This test clearly shows sites where sensory reactions are impaired, and this information can guide the clinician in determining which foods are associated with different types of the patient’s swallows.
The main advantage of FEES is that it entails direct real-time visualization of food traveling down the oropharynx and esophagus. During the FEES test, a speech language pathologist can instruct the patient to make certain physical maneuvers to find the least restrictive method of travel for the food as it passes through the throat and into the stomach. The patient can also provide feedback to the clinician in the course of the test so that the therapist can alter the volume and thickness of the food to prevent choking sensations during FEES.
Despite all of these interventions, a percutaneous endoscopic gastrostomy (PEG) tube may be needed to administer nutrition. Malnutrition, and the resulting wasting or cachexia, are grave clinical situations that may quickly occur in rapidly progressive diseases such as ALS. A similar situation may also arise in an infant or young child with SMA who cannot take in enough nutrition orally to keep up with caloric needs. In SMA, caloric needs are often greatly increased because of respiratory compromise and the increased work necessary for breathing.
PEG can readily address these issues and provide access for supplemental nutrition. Again, educating the patient and family is critical early in the disease process so they can make informed decisions regarding these issues.
Cardiac complications
Cardiac involvement may occur in most primary myopathies, including DMD, Becker muscular dystrophy (BMD), MMD, and some cases of limb-girdle muscular dystrophy (LGMD). Cardiac involvement is not seen in peripheral neuropathies or motor neuron diseases. A high (60-80%) occurrence of cardiac involvement is present in DMD and BMD patients of all ages. Dystrophin has been localized to the membrane surface of cardiac Purkinje fibers; this localization probably contributes to the cardiac conduction disturbances seen in DMD and BMD. [28, 29]
A high prevalence of abnormalities found via electrocardiography (ECG) and echocardiography exists in preadolescent patients with DMD and BMD. Nevertheless, only about 30% of patients with DMD have clinically significant cardiac complications. The myocardial impairment may remain clinically silent until the late stages of the disease, possibly because of the obligate lack of physical activity.
Pulmonary hypertension also has been implicated in the cardiorespiratory insufficiency associated with DMD. Some investigators blame congestive heart failure as the cause of death in as many as 40% of patients with DMD. [30]
An important caveat is that severe cardiac involvement in BMD occasionally precedes the clinical presentation of skeletal myopathy. Moreover, the cardiac compromise may be disproportionately severe relative to respiratory compromise in some patients with BMD. Thus, ECG and echocardiography screening are indicated at regular intervals for all patients with BMD. Patients with myocardial involvement need close follow-up and treatment by a cardiologist with expertise in this area.
Some patients with BMD may be suitable candidates for cardiac transplantation. Successful cardiac transplantation has been reported in patients with BMD with cardiac failure who were still ambulatory.
A high prevalence of abnormalities found via ECG exists in MMD. Studies have shown that about one third of patients with MMD have first-degree atrioventricular block, whereas about one fifth have left axis deviation. Only 5% have left bundle branch block. Bundle of His conduction delays have also been rarely reported. Complete heart blockage, requiring pacemaker placement, is rare but can occur. Patients with MMD should receive routine cardiac evaluations.
Pain
Pain is a significant problem for most patients with NMD, though it is not typically a direct consequence of the disease. Immobility commonly causes the pain. This may lead to adhesive capsulitis, low back pain, pressure areas on the skin, and generalized myofascial pain. Neuropathic pain is a significant problem for patients with CMT and is likely a direct consequence of the neuropathy. [31] It is now clear that pain is a common and disabling symptom in most major forms of NMD. [32, 33, 34, 35, 36, 37]
Pharmacologic pain management in patients with NMD initially includes acetaminophen (1000 mg every 6 hours). Acetaminophen may be used along with nonsteroidal anti-inflammatory drugs (NSAIDs), which may be particularly helpful if evidence indicates any active inflammatory processes (eg, joint effusion or tenosynovitis).
Tricyclic antidepressants and antiepileptic drugs are often helpful, particularly for relieving neuropathic pain. Gabapentin, an antiepileptic drug, also has the added benefit of reducing spasticity via glutamate and gamma-aminobutyric acid (GABA) pathways. Opioids may be necessary for relieving refractory pain. If required, opioids are best administered on a regular dosing schedule and titrated to the point of comfort. However, close monitoring for respiratory suppression is necessary, even in opiate-tolerant patients.
Cannabinoids, the active ingredients in marijuana, have a number of pharmacologic properties that may be applicable to the management of ALS and other NMDs. [38, 39, 40, 41, 42] Their benefits include analgesia, muscle relaxation, bronchodilation, saliva reduction, appetite stimulation, and sleep induction. In addition, they have strong antioxidative and neuroprotective effects, which may prolong neuronal cell survival. Cannabis does not suppress breathing and carries no risk of overdose, which, in this regard, makes it much safer than opiates. Further investigation into the usefulness of cannabinoids in this setting is warranted.
Nutritional management
Nutrition may be a significant problem for patients with severe NMDs, in whom obesity tends to follow shortly after the loss of functional ambulation. Obesity is common in patients with NMDs, particularly DMD, in which a prevalence of 54% has been reported. Weight control has its primary rationale in ease of care, particularly ease of transfers and skin care.
Conversely, malnutrition may mark the advanced stages of DMD, ALS, and SMA. If a severe respiratory compromise is present, the increased work of breathing may drastically increase caloric needs. To complicate the situation, this is often a time when patients lose the ability to feed themselves. A nutritionist should assess caloric requirements and construct proper dietary requirements for the patient. This should be routinely done for all patients with NMDs with a predicted FVC of less than 50% on pulmonary function testing.
PEG tube placement may facilitate nutrition by easing the intake of large amounts of calories and fluids. Patients should be reassured that they may still eat food orally for enjoyment, provided that swallowing function is intact. Another complicating factor in patients with DMD is gastroparesis, which may make feeding more difficult.
Pharmaceuticals that may improve function or prolong life
Although a comprehensive discussion of clinical trials of drugs developed to treat NMD is beyond the scope of this article, some of the major advances are worth noting. Because of its severity and rapid progression, ALS is the NMD that, to date, has received the most attention in terms of pharmaceuticals aimed at prolonging life.
Riluzole is a neuroprotective agent that appears to inhibit glutaminergic neurotransmission in the spinal cord. [43] It is the first agent approved by the US Food and Drug Administration (FDA) for use in ALS patients. Although riluzole is only modestly effective at improving life expectancy in patients with ALS, it nonetheless represents a major advance. Various neurotrophic growth factors also appear quite promising, particularly insulin-derived growth factor (myotrophin).
Data suggest that cannabidiol, a naturally occurring nonpsychotropic cannabinoid, has a potential role as a therapeutic agent for the neurodegenerative disorders produced by excessive cellular oxidation, such as ALS. This compound is chemically classified as a terpene, like tamoxifen, which also has been shown to prolong cell survival in a mouse model of ALS. This suggests a similar mechanism of action, though this remains to be delineated.
DMD, also a severe disease, has received a fair degree of study. Although not FDA-approved for this use, prednisone 1 mg/kg/day has been shown to prolong the time of ambulation in 4- to 8-year-old boys with DMD and should at least be considered for use in this disease. [44] Major adverse effects of prednisone include weight gain, osteoporosis, and mood lability. Deflazacort has similar beneficial effects and may have slightly fewer adverse effects than prednisone; however, it is not currently available in the United States.
A study showed that 200-400 mg of modafinil taken daily provided some relief from fatigue in patients with ALS. Further study is warranted before conclusive recommendations can be made. [45, 46]
New therapeutic targets have been identified that could be manipulated to mitigate sarcolemma instability, inflammation, fibrosis, and other downstream pathology. Stem cell therapies hold potential to replace diseased muscle with new, healthy tissue. Other possibilities include gene and cell-based therapies that replace, repair, or up-regulate specific genes, leading to increased production of vital proteins, thus promoting muscle growth and repair. [47, 48, 49, 50]
Antisense-mediated modulation of gene splicing with antisense oligonucleotides (AONs) has been used to restore cryptic splicing. [51, 52, 53, 54, 55, 56, 57] In Duchenne muscular dystrophy (DMD), AONs allow exon skipping, thus restoring a pathologically interrupted reading frame. This allows for the production of an internally truncated but otherwise functional, dystrophin protein. [58, 59] This therapy could potentially change the relentless progressive course of DMD into a milder, more slowly progressive course, as is seen in Becker muscular dystrophy (BMD). Once fully realized, therapeutic exon skipping could be used for a broad range of therapeutic applications in other forms of MD.
Pharmacological approaches, such as improved corticosteroid (eg, prednisone and deflazacort) dosing paradigms may slow disease progression in boys with DMD, although potential benefits may be modest. [60, 61]
Other drugs aimed at limiting free radical damage due to oxidative stress are in development, with mechanisms that include blocking pathogenic and proinflammatory stimuli. This may include inhibition of pathways such as transforming growth factor-β (TGF-β)–induced phosphorylation and tumor necrosis factor-α (TNF-α) activity, among others.
Other pharmaceutical approaches involve specific antibodies, such as MYO029, which was designed to block myostatin, an endogenous protein that normally inhibits muscle excessive growth. [60, 61] When myostatin is absent, muscles can grow much larger than normal. The Belgian Blue cow has a natural myostatin deletion mutation (a truncated myostatin gene), which disinhibits muscle growth and interferes with fat deposition, leading to huge amounts of very lean meat. [62] However, muscle growth is due primarily to hypertrophy in existing muscle cells and not to the generation of new muscle cells. Belgian Blue calves show this hypertrophic growth very early in fetal development, producing over twice the number of muscle fibers as a calf with no myostatin gene mutation. [62] This may explain why human clinical trials of MYO029 given via subcutaneous injection produced no appreciable beneficial effects in patients with facioscapulohumeral MD (FSHD), BMD, and limb-girdle MD (LGMD).
Another novel approach includes small molecular weight (< 800 Daltons) organic compounds that facilitate intracellular processes, typically through binding to a protein, nucleic acid, or polysaccharide, and altering the activity or function of the bound moiety. [63] Examples yet to be tested include molecules that act as chaperones, moving a protein or RNA segment to a place in the cell where it can be effective. Other small-molecule drugs may induce a read-through of premature stop-codon mutation, allowing for a functional protein product to be produced. [64]
Enzyme replacement therapy (ERT) has been tested in X-linked myotubular myopathy (XLMTM), a fatal congenital muscle disease caused by mutation in MTM1, the gene encoding myotubularin, a lipid phosphatase. In Mtm1-mutant mice generated by either a gene knockout, termed 1δ4, or by knock-in of a missense mutation, termed p.R69C, weakness appears largely due to impaired excitation contraction coupling. [65, 66] Short-term replacement of myotubularin with a prototypical targeted protein replacement agent (3E10Fv-MTM1) in Mtm1δ4 mice improved contractile function and muscle pathology. These promising results suggest that replacing even small amounts of myotubularin protein may improve muscle function weakness and reverse pathology.
An example of ERT approved by US regulatory authorities for use in a progressive muscle disorder called Pompe disease is alglucosidase alfa (Myozyme). Although Pompe disease is not an MD, the phenotype is very similar to LGMD. [65, 66] Pompe disease results from mutations in a gene for acid alpha-glucosidase (GAA), used by the body to break down glycogen. [62] Lysosomes ingest glycogen, which is converted by GAA into glucose to fuel muscles.
In Pompe disease, mutations in the GAA gene reduce or completely eliminate this essential enzyme, leading to excessive amounts of lysosomal glycogen, ultimately destroying the cell. The discovery of the GAA gene led to the development of a ERT. Alglucosidase alfa has been shown in clinical trials with infantile-onset patients to decrease heart size; maintain normal heart function; improve muscle function, tone, and strength; and reduce glycogen accumulation. The drug has received FDA approval for the treatment of infants and children with Pompe disease. A similar drug, alglucosidase alfa drug (Lumizyme) has been approved for late-onset (noninfantile) Pompe disease.
Rehabilitation in Neuromuscular Disease
Exercise paradigms to improve strength
Skeletal muscle weakness is the ultimate cause of most clinical problems associated with neuromuscular diseases (NMDs). A number of well-controlled studies have documented the effect of exercise as a means for NMD patients to gain strength, although much remains to be learned in this area. [67, 68]
In slowly progressive NMDs, a 12-week moderate-resistance exercise program (training at 30% of maximum isometric force) resulted in strength gains ranging from 4% to 20%, without any notable deleterious effects. However, in the same population, a 12-week high-resistance exercise program (training at the maximum weight a subject could lift 12 times) showed no benefit over the moderate-resistance program, and evidence of overwork weakness was found in some of the subjects.
In a study comparing patients who had Charcot-Marie-Tooth disease (CMT) with patients who had myotonic muscular dystrophy (MMD), the only patients who appeared to benefit significantly from a strengthening program were those with CMT; those with MMD showed neither beneficial nor detrimental effects. This clearly demonstrates that the most effective exercise regimens for patients with neuropathies and myopathies most likely vary, although further investigation is needed.
In rapidly progressive disorders like Duchenne muscular dystrophy (DMD) and amyotrophic lateral sclerosis (ALS), active ongoing muscle degeneration occurs, and the risk of overwork weakness and exercise-induced muscle injury is much greater. In this population, exercise should be prescribed with caution and a common sense approach.
Studies comparing mdx mice (mice that lack dystrophin and are genetically homologous models of DMD) with normal control mice found that in voluntary running protocols, dystrophin-deficient muscle is clearly susceptible to exercise-induced muscle injury, particularly eccentric (lengthening) muscle contractions. [69] Compared with normal mice, mdx mice show considerable avoidance behavior for exercise; this may be an intuitive survival strategy.
After ad libitum exercise on a flywheel, the extensor digitorum longus and soleus muscles of adult mdx mice significantly weakened. In addition, histochemical evidence showed considerably more damage to the exercise-weakened muscles than to the control nonexercised mdx muscles (see the image below). [70, 71]
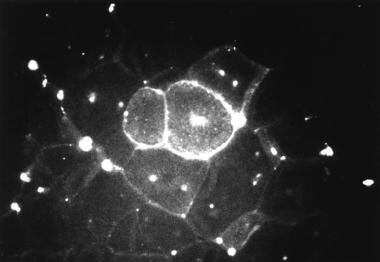 Photomicrograph of quadriceps muscle from adult mdx mouse (genetically homologous model of Duchenne muscular dystrophy) 3 days after running exercise and subsequent tail vein injection of 10,000 molecular weight fluorescent dextran. Significant intracellular staining exists in several fibers (center), indicating membrane damage. Central nuclei, present in regenerating fibers, are shown by focal intense areas of fluorescence.
Photomicrograph of quadriceps muscle from adult mdx mouse (genetically homologous model of Duchenne muscular dystrophy) 3 days after running exercise and subsequent tail vein injection of 10,000 molecular weight fluorescent dextran. Significant intracellular staining exists in several fibers (center), indicating membrane damage. Central nuclei, present in regenerating fibers, are shown by focal intense areas of fluorescence.
In view of the results from the animal studies and the available human studies, all patients with NMDs should be advised not to exercise to exhaustion, because of the risk of exercised-induced muscle damage. NMD patients in exercise programs should be monitored for signs of overwork weakness, including excessive delayed onset muscle soreness, which usually occurs 24-48 hours after exercise. Other warning signs include severe muscle cramping, heaviness in the extremities, and prolonged dyspnea.
Submaximal, low-impact aerobic exercise (walking, swimming, stationary bicycling) improves symptoms of fatigue by enhancing cardiovascular performance and increasing utilization of oxygen and substrate by muscle. This is important because fatigue is a significant limiting factor in physical performance in patients with NMDs. Fatigue in this setting is likely to be multifactorial, related to deconditioning and impaired muscular activation. [72]
Improving cardiopulmonary performance through aerobic exercise not only enhances physical functioning but also leads to a better mood state and helps fight depression. Patients with NMD have a higher frequency of depression on the Minnesota Multiphasic Personality Inventory (MMPI) than healthy populations do. Aerobic exercise also helps patients achieve and maintain ideal body weight and improves pain tolerance.
In terms of monitoring progress in an exercise program, a number of reliable, functional assessment tools are available for evaluating the effectiveness of exercise interventions, including the Timed Motor Performance assessment, which is a useful and simple measurement scale that can be used during routine clinic visits.
In the Timed Motor Performance assessment, the time required to perform selected tasks is measured in seconds with a stopwatch. Inability to complete any of the following tasks within 120 seconds is considered a failure:
-
Rise to a standing position a supine position
-
Climb 4 standard stairs (begin and end while standing with arms at sides)
-
Run or walk 30 feet (as fast as safety allows)
-
Rise to a standing position from a seated position on a chair (chair height should allow the feet to touch the floor)
-
Propel a wheelchair 30 feet
-
Put on a T-shirt (while sitting in chair)
-
Cut a 3” × 3” premarked square from a piece of paper with safety scissors (even if the lines are not followed precisely)
In terms of testing static muscle strength, manual muscle testing has been shown to be unreliable in patients with NMDs. A hand-held myometer or MicroFET type of strength-measuring device yields far more reproducible results and is just as easy to use in a clinical setting. Quantitative isokinetic strength testing, though highly reliable, requires too much sophisticated equipment to be useful in clinical settings. However, it is a good choice for research purposes.
A randomized, controlled study by Lunetta et al suggested that in patients with ALS, the use of a strictly monitored exercise program (SMEP) may significantly reduce muscle deterioration, but with no impact on length of survival. The study, which involved patients who had had definite or probable ALS for 24 months or less, found that compared with patients who were treated with 6 months of usual care, those in a 6-month SMEP scored significantly higher on the revised ALS Functional Rating Scale. However, the SMEP group did not show improvement with regard to survival, respiratory decline, or the McGill Quality of Life Questionnaire. [73]
Its many benefits notwithstanding, exercise does create some clinical problems. Muscle cramping is common in patients with NMD because of sarcolemmal instability and is often exacerbated by exercise. True muscle spasms, related to upper motor neuron spasticity, are also seen in ALS.
A number of pharmaceuticals may help treat muscle spasms. Baclofen is a good first choice because it acts via motor neuron inhibition at the spinal cord level. Tizanidine and gabapentin may also be helpful in this situation. Gabapentin would be a good choice if neuropathic pain were also present.
Benzodiazepines (or other centrally acting muscle relaxants) are not recommended because of the risk of respiratory suppression. Dantrolene is contraindicated because of its mechanism of action (impairing excitation-contraction coupling), which produces too much muscle weakness to be used in patients with NMDs.
Mexiletine is particularly useful in myotonia congenita, but cardiac conduction must be monitored while this drug is being used. Nonballistic sustained muscle stretching is also helpful and should be routinely performed after exercise.
Stretching, bracing, and surgery for contractures and scoliosis:
Joint contractures and scoliosis (see the image below) are major clinical problems in NMDs, particularly in patients with DMD and spinal muscular atrophy (SMA) type II. Consequently, a routine examination of the spine and major joints in patients with NMDs should be performed during each clinic visit.
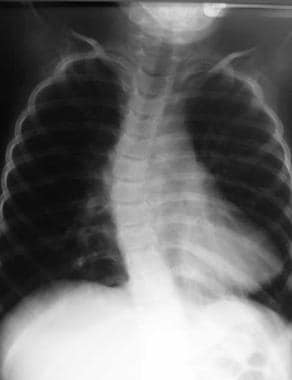 Chest radiograph from 4-year-old boy with Werdnig-Hoffmann disease (type of spinal muscular atrophy) reveals presence of significant 32° left thoracic scoliosis.
Chest radiograph from 4-year-old boy with Werdnig-Hoffmann disease (type of spinal muscular atrophy) reveals presence of significant 32° left thoracic scoliosis.
Contractures appear to be related to prolonged static limb positioning. Contractures frequently develop shortly after the patient becomes wheelchair-dependent. Several studies have documented that a lack of lower-extremity weight bearing and wheelchair dependence contribute to the development of contractures.
In ambulatory patients, upper-extremity contractures may occur and can be complicated by joint subluxation, particularly in the shoulder girdle. Slings may help provide support but will not prevent contracture formation. Again, stretching and positional splinting may slow the progression of contractures, although the actual effectiveness of this has not been well studied or documented in the literature.
Surgical release of contractures in the lower extremities may allow a patient to be functionally braced. This may prolong ambulation, although a number of studies have shown that weakness, not contracture, is the factor that makes the greatest contribution to the loss of functional ambulation.
With regard to scoliosis, there does not appear to be an etiologic relation with loss of ambulation. Although both scoliosis and wheelchair dependence are age-related, several studies have found there to be no relation between the 2 phenomena. A large study by Lord et al reported an almost 4-year difference between wheelchair dependence and the onset of significant scoliosis in patients with DMD. [74]
Indeed, many DMD and SMA II patients develop scoliosis before becoming wheelchair-dependent. Disease progression with increasing weakness of trunk musculature is more likely the major underlying cause of neuromuscular scoliosis. However, trunk flexor or extensor strength is hard to measure quantitatively. In NMD patients, scoliosis did not appear to correlate with trunk strength on manual muscle testing (though this method cannot measure asymmetric strength and is not a reliable strength-assessment method in this population).
Patients with DMD typically develop scoliosis around the time of the adolescent growth spurt. [75] However, patients with SMA II may develop scoliosis much earlier. In DMD and SMA, studies have shown that thoracolumbar curves are much more common than lumbar curves. Patients with DMD and SMA should be monitored closely with serial radiographs because the curve may suddenly progress.
Spinal bracing has not been shown to be effective in preventing progression of neuromuscular scoliosis (though most studies involved patients with SMA). Thus, spinal instrumentation and fusion is the only treatment option known to be effective. This should be performed before the primary curve exceeds 25° and FVC has not dropped below 50% of predicted. Complications substantially increase if the patient already has compromised breathing.
Unfortunately, correction of the scoliosis with fusion does not appear to improve pulmonary function. [76] Nonetheless, fusion does improve quality of life (QOL) by facilitating positioning, seating, and transfers. If the curve progresses much beyond 40°, successful correction via fusion is much less likely.
For the extremities, the goals of bracing should be to improve function and joint stability. Long leg bracing to prolong ambulation time in DMD has been one of the better-studied uses of bracing in NMDs. A number of studies have shown that ambulatory ability may be prolonged by up to 2 years with the use of long leg braces and appropriate contracture release. However, whether this represents a subset of patients with a slower disease progression and relatively less weakness is unclear.
Furthermore, no clear association exists between prolonging ambulation with long leg bracing and delaying or decreasing scoliosis in patients with DMD. If bracing is used, a long leg brace or a knee-ankle-foot orthosis is generally needed because of the amount of weakness in hip and knee extension, as well as in ankle plantar flexion and dorsiflexion.
Most patients with CMT require short leg braces or ankle-foot orthoses. Ideally, these should be custom-made with a lightweight polymer (polypropylene or carbon fiber). They should fit intimately to prevent skin problems and to provide good stability. If a pressure sore occurs, the brace should be removed until the patient heals.
Double metal upright ankle-foot orthoses may be built into the shoe but are usually too heavy and may limit ambulation. If the ankle is significantly unstable, the braces should be high-profile (ie, should come around in front of the malleoli). Pes cavus and hammertoe deformities can be accommodated with built-up arches and metatarsal bars.
Patients with CMT and other sensory neuropathies are at very high risk for skin ulcers and neuropathic arthritis (Charcot joint). Thus, skin integrity and joint stability should be checked during every clinic visit.
Any patient with weakness due to an NMD may benefit from bracing, depending on the distribution of weakness, gait problems, and joint instability. The decision to brace should include the risk of the brace’s added weight and the willingness of the patient to use the brace. After being fitted with braces, patients with NMDs should be referred for a course of physical therapy to help them learn to use the devices effectively.
Vocational, psychosocial, and quality-of-life issues
Patients with NMDs, particularly those with advanced ALS and DMD, may experience reactive clinical depression. As noted (see above), studies involving patients with NMDs have shown elevated scores for depression on MMPI testing. In one study, depression was more closely associated with the level of independent functioning than with limb strength. Thus, good family, social, and religious support systems are critical.
The possibility that other family members and caregivers may also become depressed should not be overlooked. Group and family counseling may be beneficial. Patients with NMDs should be referred to support groups, which are also excellent resources for psychological support and problem solving. If necessary, a patient should be referred to a mental health professional. [77, 78]
Antidepressant medicine may also help with mood elevation and may improve appetite and sleep. In patients with ALS, tricyclic antidepressants with significant anticholinergic activity dry up oral secretions. This can help minimize drooling.
Significant cognitive involvement is common in patients with MMD and some congenital mitochondrial myopathies. Learning disabilities are also seen in about one third of boys with DMD. Beyond that, most people with NMD show normal intelligence. Unfortunately, employment rates for people with NMDs are significantly lower than for the able-bodied population. In the NMD population, a higher level of education correlated more closely with employment rate than with functional level or physical performance.
The self-esteem levels noted on personality testing also correlated positively with the level of education and employment. This implies that altered personality profiles in patients with NMDs may have a significant effect on their ability to integrate into mainstream society and to hold steady employment rates. In this regard, education appears to be at least as important as physical abilities with respect to employability and self-esteem in people with NMDs.
NMD patients are incredibly diverse, and this may be why very few studies have attempted to determine the effect of NMDs on QOL. The studies that have been done were criticized for lumping together many differing NMDs in a poorly defined study group and for mainly using generic instruments to define QOL. Thus, these studies rarely used clinically meaningful data on physical and emotional functioning; they focused on the extreme manifestations (eg, severe pain) rather than looking at how patients experience and deal with their NMD.
Some disease-specific QOL assessment tools, most notably the NeuroQOL and the Amyotrophic Lateral Sclerosis Assessment Questionnaire (ALSAQ-40), have been developed specifically for NMD patients. These disease-specific measures are especially useful for evaluating patients’ QOL and psychosocial functioning as their NMD progresses. Beyond ALS, there has been a dearth of studies using QOL measurements to study NMDs. This is an area where further investigation is strongly warranted.
Equipment
Proper equipment can significantly improve QOL for NMD patients. Common examples of equipment include hospital beds, commode chairs, wheelchairs and wheelchair ramps, handheld showers, bathtub benches, grab bars, raised toilet seats, and many others. An occupational therapist is best qualified to help determine whether any of these devices would be useful for a patient with an NMD.
Wheelchairs are a critical component of mobility for people with NMDs. They must be fitted with the appropriate frame size, type of seat, lumbar support, and cushioning to avoid pressure ulcers. They should be equipped with other mechanical accessorial devices (eg, tilt ability) to provide comfort and to protect the skin. A physical or occupational therapist should evaluate the patient to ensure a proper wheelchair prescription. Patients who are simply given a prescription for a wheelchair often get a chair that fits poorly or lacks the proper components.
Power wheelchairs are indicated for most patients with NMDs who can no longer ambulate. These patients do not have enough upper-extremity strength to propel a manual chair by themselves. Although power wheelchairs are expensive, they can be justified to third-party payers on the grounds that they help prolong independent mobility, thus decreasing medical and psychological comorbidity.
In patients who can still ambulate, walkers or quad (4-point) canes help reduce the risk of falling. Pressure-relieving mattresses, along with foam wedges for proper positioning, help prevent pressure skin ulcers. In some patients with NMDs, particularly ALS, severe weakness in the neck musculature causes neck pain and muscle spasms. A cervical collar, particularly the Freeman or Headmaster type (a wire-frame collar with padding over the pressure points), may be very helpful.
In patients with dysarthria (typically patients with ALS), augmentative communicative aids, including an alphabet board, a word board, or a computer-based speech synthesizer, can be very helpful for maintaining functional communication. A speech language pathologist is best qualified to determine which, if any, of these devices would work best.
Future Research
Treatment advances
The past decade has seen advances in understanding the molecular genetic basis and pathophysiology of neuromuscular disorders (NMDs), including the major forms of muscular dystrophy (MD). [32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54]
Research into both the molecular basis and pathophysiology of many forms of MD has led to strategies aimed at correcting primary cellular deficiencies. Not only do we have a better functional understanding of cellular deficits and abnormal pathophysiological responses in dystrophic muscle, but we also have new technologies such as regenerative medicine and genetic manipulation, which hold real potential for curative treatments.
Studies using isolated muscle preparations such as skinned single fibers allow for accurate comparison of stretch-induced force deficits in normal and dystrophic muscle. [33] It is also now possible to monitor cellular processes such as light-chain phosphorylation in response to eccentric contraction-induced injury. [34]
New technologies also allow for more accurate measurements in MD animal models, such as studying the kinematics of gait, as well as myofiber injury and regeneration, in golden retrievers with MD. [35, 36] New data are forthcoming about the effects of aging in dystrophic muscle. [38, 39]
-
Werdnig-Hoffman disease (type of spinal muscular atrophy). Small muscle fibers within separate muscle fascicles.
-
Axial magnetic resonance imaging (MRI) of calves, demonstrating significant atrophy and increased signal intensity (arrows) throughout anterior and posterior compartment muscles in right calf, as compared with left calf, in patient with focal motor neuron disease.
-
Kugelberg-Welander disease (type of spinal muscular atrophy). Marked variation in muscle fiber size, along with increased perimysial connective tissue.
-
Foot deformities in 16-year-old boy with Charcot-Marie-Tooth disease type 1A.
-
(A) Normal dystrophin staining. (B) Intermediate dystrophin staining in patient with Becker muscular dystrophy. (C) Absent dystrophin staining in patient with Duchenne muscular dystrophy.
-
Photomicrograph of quadriceps muscle from adult mdx mouse (genetically homologous model of Duchenne muscular dystrophy) 3 days after running exercise and subsequent tail vein injection of 10,000 molecular weight fluorescent dextran. Significant intracellular staining exists in several fibers (center), indicating membrane damage. Central nuclei, present in regenerating fibers, are shown by focal intense areas of fluorescence.
-
Chest radiograph from 4-year-old boy with Werdnig-Hoffmann disease (type of spinal muscular atrophy) reveals presence of significant 32° left thoracic scoliosis.







