Recovery Considerations
Stroke rehabilitation is a combined and coordinated use of medical, social, educational, and vocational measures to retrain a person who has suffered a stroke to his/her maximal physical, psychological, social, and vocational potential, consistent with physiologic and environmental limitations. The cellular mechanisms behind stroke are seen in the image below.
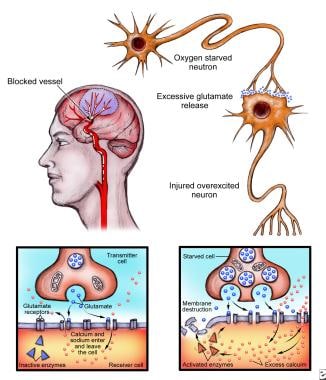 When the brain suffers an injury, such as a stroke, neurons release glutamate onto nearby neurons, which become excited and overloaded with calcium, after which they die (left). Normal neurotransmission (above) is altered during injury, causing excess calcium to activate enzymes, eventually leading to destruction of the cell. Since this process occurs via glutamate receptors, including N-Methyl-D-aspartate (NMDA) receptors, scientists believe that damage can be stopped through the use of agents that block these receptors.
When the brain suffers an injury, such as a stroke, neurons release glutamate onto nearby neurons, which become excited and overloaded with calcium, after which they die (left). Normal neurotransmission (above) is altered during injury, causing excess calcium to activate enzymes, eventually leading to destruction of the cell. Since this process occurs via glutamate receptors, including N-Methyl-D-aspartate (NMDA) receptors, scientists believe that damage can be stopped through the use of agents that block these receptors.
Evidence from clinical trials supports the premise that early initiation of therapy favorably influences recovery from stroke. When the initiation of therapy is delayed, patients may in the interim develop avoidable secondary complications, such as contractures and deconditioning.
In addition, many studies show that stroke rehabilitation can improve functional ability even in patients who are elderly or medically ill and who have severe neurologic and functional deficits.
The initial clinical examination of a patient with an acute stroke includes a thorough, detailed neurologic examination. The neurologic findings are used by the rehabilitation team for prognostication, development of the specific details of the rehabilitation plan, and selection of the appropriate setting for rehabilitation.
Reassessment of the patient's condition during rehabilitation provides a means of monitoring progress and subsequently evaluating outcome. The initial rehabilitation assessment should begin immediately following onset, within 2-7 days, and then subsequently at repeated intervals.
Go to Stroke, Ischemic, for more complete information on this topic.
Timing, extent, and types of recovery
Patients recover after stroke in 2 different, but related, ways.
A reduction in the extent of neurologic impairment can result from spontaneous, natural neurologic recovery (via the effects of treatments that limit the extent of the stroke) or from other interventions that enhance neurologic functioning. A patient demonstrating this form of recovery presents with improvements in motor control, language ability, or other primary neurologic functions.
The second type of recovery demonstrated by stroke patients is the improved ability to perform daily functions within the limitations of their physical impairments. A patient who has sensorimotor, cognitive, or behavioral deficits resulting from stroke may regain the capacity to carry out activities of daily living (ADL), such as feeding himself/herself, dressing, bathing, and toileting, even if some degree of residual physical impairment remains.
The ability to perform these tasks can improve through adaptation and training in the presence or absence of natural neurologic recovery, which is thought to be the element of recovery on which rehabilitation exerts the greatest effect.
Hemiparesis and motor recovery have been the most studied of all stroke impairments. As many as 88% of patients with acute stroke have hemiparesis.
In a classic report, Twitchell described in detail the pattern of motor recovery following stroke. [1] At onset, the upper extremity (UE) is more involved than the lower extremity (LE), and eventual motor recovery in the UE is less than in the LE. The severity of UE weakness at onset and the timing of the return of movement in the hand are important predictors of eventual motor recovery in the UE. A systematic review of 58 studies confirms the most important predictive factor for upper limb recovery following stroke is the initial severity of motor impairment or function. [2] The prognosis for return of useful hand function is unfavorable when UE paralysis is complete at onset or grasp strength is not measurable by 4 weeks.
However, as many as 9% of patients with severe UE weakness at onset may gain good recovery of hand function. As many as 70% of patients showing some motor recovery in the hand by 4 weeks make a full or good recovery. Full recovery, when it occurs, usually is complete within 3 months of onset.
Bard and Hirshberg asserted that if no initial motion is noticed during the first 3 weeks or if motion in one segment is not followed within a week by the appearance of motion in a second segment, the prognosis for recovery of full motion is not favorable.
Although most recovery from stroke takes place in the first 3 months, and only minor additional measurable improvement occurs after the 6 months following onset, recovery may continue over a longer period of time in some patients who have significant partial return of voluntary movement.
Criteria for admission to a comprehensive rehabilitation program
Criteria for a patient’s admission to a comprehensive rehabilitation program may include the following:
-
Stable neurologic status
-
Significant persisting neurologic deficit
-
Identified disability affecting at least 2 of 5 functions, including mobility, self-care activities, communication, bowel or bladder control, and swallowing
-
Sufficient cognitive function to learn
-
Sufficient communicative ability to engage with therapists
-
Physical ability to tolerate the active program
-
Achievable therapeutic goals
Theories of Recovery
One theory of motor recovery is that collateral sprouting from intact cells to the denervated region occurs after some or all input has been destroyed.
Another theory suggests that there is an unmasking of neural pathways and synapses that are not normally used but that can be called upon when the dominant system fails (excitability to capture effects of remaining input).
Mechanisms of Recovery
The first recovery mechanism is resolution of harmful local factors, which generally accounts for early spontaneous improvement after stroke (usually within the first 3-6 mo). These processes include resolution of local edema, resorption of local toxins, improvement of local circulation, and recovery of partially damaged ischemic neurons.
Neuroplasticity
The second recovery mechanism, which may continue for many months, is neuroplasticity, which can take place early or late. Brain plasticity is the ability of the nervous system to modify its structural and functional organization. The 2 most plausible forms of plasticity are collateral sprouting of new synaptic connections and unmasking of previously latent functional pathways.
Other mechanisms of plasticity include assumption of function by undamaged, redundant neural pathways, reversibility from diaschisis, denervation supersensitivity, and regenerative proximal sprouting of transected neuronal axons. Experimental evidence indicates that plasticity can be altered by several external factors, including pharmacologic agents, electrical stimulation, and environmental stimulation.
A key aspect of neuroplasticity that has important implications for rehabilitation is the fact that the modifications in neuronal networks are use-dependent. Animal experimental studies and clinical trials in humans have shown that forced use and functional training contribute to improved function. On the other hand, techniques that promote nonuse may inhibit recovery.
In the past, the conventional wisdom was that benefits from rehabilitation were achieved primarily through training patients in new techniques that compensate for impairments (for example, using the uninvolved hand to achieve self-care independence). This approach avoided intense therapy on the weak upper limb.
Currently, it is recognized that repeated participation by patients in active physical therapeutic programs probably provides direct influence on the process of functional reorganization in the brain and enhances neurologic recovery.
Pattern of Disability
After stroke occurs, total loss of voluntary movement may be noted in involved extremities, with loss or decrease in muscle stretch reflexes (MSRs). Within 48 hours, MSRs and finger jerks are more active on the involved side, although they may require 3-29 days to develop. Within a short period, tone appears in the wrist and finger flexors, as well as in the ankle plantar flexors. As a result, the UE is prone to demonstrate the adductor/flexor pattern, and the LE is prone to demonstrate the adductor/extensor pattern.
Development of spasticity
In 1-30 days, spasticity appears, resulting in resting posture. In the upper extremity, this posture takes the following form:
-
Shoulder - In adduction and internal rotation
-
Elbow - In flexion
-
Forearm - In pronation/supination
-
Wrist and fingers - In flexion
In the lower extremity, resting posture develops as follows:
-
Hip - In adduction and extension
-
Knee - In extension
-
Ankle - In plantar flexion
-
Foot - In inversion
Within 1-38 days after stroke, clonus appears in ankle plantar flexors, and the onset of clasp-knife phenomenon occurs within 3-31 days.
The strength of the ankle dorsiflexors plays a large role in determining walking speed following stroke. [3]
Spasticity in the lower extremity decreases with increased volitional movement, but MSRs always remain increased, despite total recovery.
Pattern of Recovery
Recovery of function in the UEs
Recovery of UE flexor synergy occurs as follows:
-
Shoulder flexion - 6-33 days
-
Elbow flexion - 1-6 days later
-
Finger and wrist flexion - 1-13 days later
-
Shoulder adduction/internal rotation
Clinically, flexor synergy can also present as follows:
-
Scapula retraction/elevation
-
Shoulder abduction (90°)/external rotation
-
Elbow flexion (acute angle)
-
Forearm supination (full range)
Recovery of UE extensor synergy occurs as follows:
-
Shoulder
-
Elbow
-
Wrist/finger extension
Clinically, extensor synergy presents as follows:
-
Scapula protraction
-
Humerus flexion/internal rotation
-
Elbow extension
-
Forearm pronation
In a study of 188 patients with stroke, Nijland et al found that assessment of finger extension and shoulder abduction within 72 hours after stroke can help to predict upper limb recovery. If, by the second day following stroke, patients in whom upper limb motor function was affected were capable of some voluntary extension of the fingers and some abduction of the hemiplegic shoulder, there was a 0.98 probability that they would regain some dexterity by 6 months. [4]
Patients with no such voluntary movement on the second day, according to the study, had only a 0.25 probability of regaining dexterity by 6 months. Full recovery at 6 months was achieved in 60% of patients with some early finger extension.
Recovery of function in the LEs
Recovery of LE flexor synergy occurs as follows:
-
Hip flexion/adduction - 1-31 days
-
Knee flexion - 1-2 days later
-
Ankle/toe dorsiflexion - 25-90 days
LE extensor synergy is recovered first in hip/knee extension and then in ankle plantar flexion.
PT Options in Stroke
Rehabilitation should include physical therapy (PT) that is directed at specific training of skills and at functional training. [5] Therapy should be given with sufficient intensity to promote skill acquisition. Major theories of rehabilitation training include the following:
-
Traditional therapy
-
Bobath concept: Neurodevelopmental training
-
Proprioceptive neuromuscular facilitation
-
Brunnstrom
-
Sensorimotor therapy: Rood approach
-
Motor relearning program: Carr approach
-
Constraint-induced movement therapy (CIMT)
-
Functional electrical stimulation (FES)
-
Electromyographic biofeedback (EMG-BF)
-
Robotic devices
Traditional therapy
This form of therapy employs range-of-motion (ROM), strengthening, mobilization, and compensatory techniques. The process of mental practice may also be used to improve the performance of certain activities. [6] This is when a patient mentally rehearses an action without physically performing the action. Current evidence is not clear on whether this practice, in conjunction with physical practice, actually improves motor capacity of the upper limb region. Further studies are required. [7]
Bobath concept
According to the Bobath concept, probably the most commonly used approach, muscle patterns, not isolated movements, are utilized for motion. The theory states that persons with motor deficiencies following stroke are unable to direct nervous impulses to muscles in the different combinations used by persons with an intact central nervous system (CNS).
The therapy, therefore, is meant to suppress abnormal muscle patterns before normal patterns are introduced. Abnormal patterns are modified at proximal key points of control, such as the neck, spine, shoulder, and pelvis.
Proprioceptive neuromuscular facilitation
This form of therapy aims to stimulate nerve/muscle/sensory receptors to evoke response through manual stimuli to increase ease of movement and promote function.
Brunnstrom movement therapy
This therapy involves central facilitation using Twitchell's recovery. It aims to enhance specific synergies through the use of cutaneous/proprioceptive stimuli.
Sensorimotor therapy
This therapy uses cutaneous sensorimotor stimulation to modify muscle tone and voluntary activity.
Motor relearning program
This therapy uses cognitive motor relearning theory to allow the patient to move functionally; it teaches general strategies for solving motor problems.
Studies
Every patient should avoid strenuous exercise after stroke, but it is a good idea to participate in an individualized exercise program. At 1 year poststroke, improvement in functional walking ability was seen in stroke patients who underwent either locomotor training, including body weight supported treadmill, or a progressive home exercise program supervised by a physical therapist. No significant differences in improvement were found between the 2 groups. [8] Reports in the literature stated that for young stroke survivors who participated in an aerobic fitness program, improvement in fitness levels, ambulatory speed, and life satisfaction was statistically significant.
A study by Kawakami et al indicated that a higher amount of physical therapy (4.5-6 exercise sessions per day [86 patients] vs. 2.5-3 daily sessions [91 patients]) leads to greater improvements in walking ability 4 weeks after hospital admission and at discharge, in patients with severe or complete poststroke lower limb paralysis. (However, the study did not find such differences in improvement in those with mild or moderate poststroke paralysis.) [9]
Similarly, a retrospective study by Kimura et al indicated that in patients with subacute stroke, the administration, along with conventional rehabilitation therapy, of additional physical and occupational therapy leads to better post-stroke functional recovery. Patients who underwent physical and occupational therapy only on weekdays had a mean Functional Independence Measure (FIM) effectiveness score of 39.3, compared with 43.4 and 54.3 for individuals who also had physical therapy or physical and occupational therapy, respectively, on weekends. [10]
A study by Persson et al found that patients with good postural control in the first week following the onset of stroke tended to report higher physical activity levels in the first year poststroke. The study included 96 patients, who were followed up after a first-ever stroke. [11]
Results from a randomized, controlled, assessor-blinded study indicated that even long after a stroke, kinesthetic ability training, administered in combination with a conventional rehabilitation program, can improve balance in hemiparetic stroke patients. [12]
The inclusion of breathing retraining (BRT) and inspiratory muscle training (IMT) in the rehabilitation program of patients who have suffered a stroke can result in improved respiratory muscle function, exercise capacity, and quality of life, according to a study by Sutbeyaz et al. In this study, patients received BRT and IMT training for half an hour daily, 6 times a week for 6 weeks. [13]
The Extremity Constraint Induced Therapy Evaluation (EXCITE) trial indicated that patients undergoing a 2-week CIMT program can achieve significant improvements in arm motor function that last for at least 1 year. The study involved 220 patients with predominantly ischemic stroke, 106 of whom underwent 2 weeks of CIMT and the rest of whom were treated with usual and customary care, with the aim of improving upper extremity function. Patients in the study had suffered a first stroke 3-9 months prior to treatment. Over a 12-month follow-up period, improvement in the Wolf Motor Function Test (WMFT) Performance Time, as well as on the Motor Activity Log (MAL) Amount of Use and Quality of Movement scales, was greater in the CIMT patients than in the control group. [14]
Similarly, results from a systematic review indicated that modified CIMT is more effective than traditional rehabilitation in reducing a patient's disability level, being able to improve upper extremity ability and increase movement spontaneity. [15] Further studies are needed on CIMT’s effectiveness in kinematic analysis.
In a randomized, clinical pilot study with a 6-month follow-up, the practicality and efficacy of conventional neurologic therapy, constraint-induced therapy, and therapeutic climbing to improve minimal to moderate arm and hand function in stroke patients was evaluated. The study concluded that improvement of arm and hand function in the intermediate term was best achieved using the constraint-induced therapy approach. [16]
A randomized clinical trial by Terranova et al indicated that in patients with chronic stroke, results from upper limb motor rehabilitation do not significantly differ between robot-assisted therapy (RT) and CIMT. Patients in the study underwent RT or CIMT, in combination with a conventional rehabilitation program. Results, as measured using the Wolf Motor Function Test and the Fugl-Meyer Assessment-upper limb, were comparable, with both types of rehabilitation yielding significant functional improvements. [17]
Occupational Therapy in Stroke
Most patients with significant neurologic impairment who survive a stroke are dependent on others for performance of basic ADL (ie, bathing, dressing, feeding, toileting, grooming, transfers). The capacity of individuals to perform these activities usually is scored on disability rating scales, such as the Functional Independence Measure. Almost all patients show improved performance of ADL as recovery occurs.
Most improvement is noted in the first 6 months, although as many as 5% of patients show continued measurable improvement up to 12 months postonset. Other patients may show some functional improvement beyond 6 months, even though the disability scales usually fail to detect further improvement because of their limited sensitivity at the upper end of the functional range.
Reports of the levels of functional independence eventually reached by stroke patients after recovery vary from one author to another. This variability probably reflects differences between study populations, methods of treatment, follow-up, and data reporting. In most reports, 47-76% of patients achieve partial or total independence in the performance of ADL.
Most authors who have attempted to determine which factors predict ultimate ADL functional outcome have used multivariate analysis. Of the many independent variables tested, those listed below have been reported to have the most influence on outcome. However, not all of these factors have been shown to predict outcome status statistically in every study. Factors predicting poor ADL outcome include the following:
-
Advanced age
-
Comorbidities
-
Myocardial infarction
-
Diabetes mellitus
-
Severe stroke
-
Severe weakness
-
Poor sitting balance
-
Visuospatial deficits
-
Mental changes
-
Incontinence
-
Low initial ADL scores
-
Delay in initiating rehabilitation following onset
Aphasia Therapy
Approximately one third of patients with acute stroke have clinical features of aphasia. Language function in many of these patients improves, and, at 6 months or more after stroke, only 12-18% of patients have identifiable aphasia.
The most improvement in patients with aphasia occurs in the first 2-3 months, with the recovery rate dropping significantly after 6 months. Skilbeck and colleagues, however, reported that patients with aphasia continue to show some late improvement in language function even more than 1 year after onset.
Patients who are classified initially as having Broca aphasia have variable outcomes. In patients with large hemisphere lesions, Broca aphasia persists with little recovery. Patients with smaller lesions confined to the posterior frontal lobe often show early progressive improvement, but the impairment may evolve into a milder form of aphasia with anomia and difficulty finding words. Patients with global aphasia tend to progress slowly, with comprehension often improving more than expressive ability does.
The communicative ability of patients who initially have global aphasia improves over a longer period of time, up to a year or more postonset. Patients with global aphasia associated with large lesions may show only minor recovery, but recovery may be quite good in patients with smaller lesions. The extent of language recovery associated with Wernicke aphasia is variable.
Associated Conditions
Most patients with stroke who undergo rehabilitation have many other associated medical conditions that require professional attention. These problems might be preexisting medical illnesses that necessitate ongoing care (eg, hypertension, diabetes mellitus [DM]), secondary poststroke complications (eg, deep venous thrombosis, pneumonia), or acute poststroke exacerbations of preexisting chronic diseases (such as angina in a patient with ischemic heart disease).
Management of these conditions can constitute major portions of the rehabilitation effort. Some patients may be more disabled by certain associated comorbid diseases than by the stroke itself.
The occurrence of these associated conditions has several implications for management of stroke cases during and after rehabilitation. First, these problems can detract from the benefits of rehabilitation. Some medical problems, such as heart disease, have been found to affect the course and outcome of rehabilitation adversely following a stroke. Intercurrent medical complications can limit the patient's ability to participate in therapeutic exercise programs, inhibit functional skill performance, and reduce the likelihood of achieving favorable outcomes from rehabilitation.
The rehabilitation interventions also might affect the medical condition adversely, causing an exacerbation of the disease or necessitating an adjustment in the treatment program. Patients who are treated in a stroke unit have better outcomes at discharge than do patients who are not. [18]
A retrospective, observational study assessed the factors that help decide the postacute level of care for stroke patients with aspiration pneumonia (ASPNA). It concluded that patients with ASPNA and a National Institutes of Health Stroke Scale (NIHSS) value of 7.44 or greater showed the need for additional postacute care. Those patients with ASPNA and a NIHSS value of 10.93 or greater showed the need for a skilled nursing facility or subacute care. Patients older than 69 years with ASPNA had increased chances of placement in subacute care. [19]
Surgical Options
Tendon release can be performed in cases of severe spasticity or contractures.
Carotid endarterectomy can be carried out in patients with stenosis of 70% or greater.
There is no longer any clear indication for carotid artery bypass to prevent stroke or in patients who have had a TIA. No benefit has been demonstrated from the surgery.
Although there have been reports of successful cases involving surgical bypass or endarterectomy involving the posterior circulation, these procedures remain largely experimental.
Consultations in Stroke
Consultations with neurologists and physiatrists are important aspects of treatment in patients who have suffered stroke.
Consultations with psychologists are also essential. Psychosocial issues obviously are very important in cases of stroke. Numerous studies have reported on the influence of the psychological adjustment and coping mechanisms of the patient, as well as those of his/her spouse and other family members, in determining the patient’s outcome.
Other Treatment Options
Biofeedback attempts to modify autonomic functions, pain, and motor disturbances through acquired volitional control, using auditory, visual, and sensory clues. [20]
Functional electrical stimulation commonly is employed in the UEs and LEs to improve strength, encourage and augment early active ROM, assist in the management of dependent peripheral edema through forceful isotonic muscle contraction, and establish early proprioceptive joint sense in the sensory-compromised patient. [21]
Rehabilitation programs are offered in different settings, such as acute inpatient rehabilitation units, subacute inpatient rehabilitation units, home care environments, and outpatient centers. The acute rehabilitation setting is appropriate for patients who meet the admission criteria and are able to tolerate 3 hours or more of active therapy per day.
An acute rehabilitation setting is preferred if the patient requires close monitoring of his/her medical status by medical and nursing professionals. If the patient's medical status is stable but the patient is unable to tolerate more than 1 hour of therapy a day, a subacute rehabilitation or skilled nursing setting is more appropriate. Patients who are independent or require only minimal assistance in self-care tasks and mobility are suited for outpatient therapy or a home care program.
Rehabilitation units
Medical stability traditionally has been required for admission of a patient to a specialized rehabilitation unit; however, hospitals increasingly are transferring patients from acute wards to rehabilitation units at earlier stages, often when the patients still have unresolved medical problems.
This practice has forced rehabilitation centers to expand their resources to care for these more complex cases and to provide closer medical and nursing monitoring. Local institutional referral patterns and practices usually determine the timing of transfer, but if earlier transfer to rehabilitation can be accomplished safely, patient care may be enhanced by earlier active participation of the patient in the rehabilitation program. [22]
Planning for discharge from the inpatient rehabilitation program should begin on admission. Discharge functional status, destination of discharge, and length of hospital stay are comparable in patients with a good prognosis. Discharge functional status is comparable in patients with an unfavorable prognosis, but mortality is higher and the hospital stay is longer in medical wards.
Discharge from the hospital often is thought of as the end of rehabilitation, with the assumption that a good program prepares the patient for reintegration into the home and community; however, hospital discharge instead should be looked at as the end of the beginning of a new life in which the patient faces the challenge of adapting to different roles and relationships and of searching for new meaning in life.
This adaptation involves resuming former roles in the family and with friends as much as possible and finding ways to live a meaningful life in the community.
Postacute rehabilitation
During postacute rehabilitation, all patients should be monitored carefully for evidence of cardiac disease. The classic features of coronary artery disease and congestive heart failure may be present, but often they are not. Ischemia may be silent. [23]
The clinical clues to significant coexisting heart disease may be subtle (eg, slower than expected progress, excessive fatigue, lethargy, mental changes). These cardiac complications can be treated successfully and are not contraindications to rehabilitation. The patient should undergo appropriate cardiac investigation with electrocardiography, Holter monitoring, and echocardiography and also should receive optimal therapy.
Early initiation of therapies is desirable. Beginning rehabilitation early minimizes secondary complications, such as contractures and deconditioning, and helps to motivate the patient. Whether more intense therapy as an independent variable improves ultimate functional recovery is not known. [24]
Evaluation of neurologic impairments should be made repeatedly during the course of the rehabilitation program. Ideally, evaluation should be made weekly in the early phases of rehabilitation to allow monitoring of the recovery process and to guide the therapeutic intervention. A clear need for committed medical direction is evident in patients who have sustained strokes.
The role of the clinician includes provision of medical care. Many patients have ongoing associated medical problems that require appropriate monitoring and therapy. The clinician must act as a medical counselor, offering reasonable prognostication to patient and family, along with guidance in reduction of stroke risk factors and ongoing medical care. The clinician also must give leadership to the team and assist in developing treatment protocols and setting treatment expectations.
The multiple problems that a patient can have following stroke require the active participation of a team of professionals. The treatment activities of the team members must be coordinated so that detailed evaluations are shared and agreements made regarding goals and treatment interventions.
Each of the professional therapists on the team should be knowledgeable about the appropriate interventions within his/her discipline for treating the disabilities of patients following stroke. The interventions should be directed at achieving specific therapeutic goals, which may be for the short term (for example, weekly goals) or longer term (for instance, goals to be reached by discharge). Having achieved those goals, the patient moves on to the next phase of rehabilitation or is discharged home to continue treatment as an outpatient.
Rehabilitation requires a functional approach. When impairments cannot be altered, every effort should be made to assist patients in compensating for deficits and adapting to alternative methods so that they can achieve optimal functional independence.
Home Care in Stroke
A study by Young and Forster found home care to be cheaper than day hospital services (£385 vs £620 [approximately $546 vs $880]). [25]
Outcome measurements have indicated a modest advantage in favor of home care.
No difference in outcome was found between home care and hospital-based rehabilitation following acute care.
A 2012 Cochrane review found insufficient evidence to make recommendations about the effects of home-based therapy programs versus placebo, no intervention or usual care for upper limb recovery in stroke. [26]
Hospital-based services are 27% more expensive than home care services.
Geriatric ward patients are 2.4 times less likely to die or to become institutionalized by 6 months if placed in day hospital service.
Stroke unit patients demonstrate superior ADL performance at 6 months with home care (2.6 times more expensive) than they do with outpatient therapy.
General medical ward patients had similar outcomes, although outpatient services cost 56% of home care.
Home care risks
Risks for suboptimal home care (72.6% prediction/validation rate) include the following:
-
A depressed caregiver
-
Inadequate knowledge of how to care for a family member following a stroke
-
A dysfunctional family
Cardiac Precautions
The rehabilitation management of patients with identified cardiac complications should include formal clinical monitoring of pulse and blood pressure during physical activities. Brief electrocardiac monitoring during exercise can add more specific information.
Note that in deconditioned patients, the resting heart rate may be high, and, in an elderly patient, the estimated limit for heart rate based on 50% above resting may be too high. For patients on beta blockers, a reasonable limit might be a heart rate of around 20 beats above the resting level.
A useful set of cardiac precautions in patients undergoing rehabilitation was developed by Fletcher and colleagues. Activity should be terminated if any of the following symptoms develop:
-
New onset of cardiopulmonary symptoms
-
Heart rate decreases to less than 20% of baseline
-
Heart rate increases to greater than 50% of baseline
-
Systolic BP increases to 240 mm Hg
-
Systolic BP decreases 30 mm Hg from baseline or to less than 90 mm Hg
-
Diastolic shortening fraction increases to 120 mm Hg
Complications During Rehabilitation
Medical complications frequently occur during the postacute phase of rehabilitation, affecting up to 60% of patients (and up to 94% of patients with severe lesions).
Common medical complications include the following:
-
Pulmonary aspiration, pneumonia - 40%
-
Urinary tract infection - 40%
-
Depression - 30%
-
Musculoskeletal pain, reflux sympathetic dystrophy - 30%
-
Falls - 25%
-
Malnutrition - 16%
-
Venous thromboembolism - 6%
-
Pressure ulcer - 3%
The means of treating depression in patients following stroke remains uncertain. One study found evidence that pharmacotherapy can reduce depressive symptoms in these patients but that it can also increase adverse events. [27] The report found no evidence that psychotherapy reduces depression.
Common neurologic complications include the following:
-
Toxic or metabolic encephalopathy - 10%
-
Stroke progression - 5%
-
Seizures - 4%
In ischemic stroke patients who were followed over the course of 2-4 years, seizures developed in 6-9% of patients. Seizures developed in 26% of patients with cortical lesions and in 2% of patients with subcortical lesions. Risk factors include the following:
-
Lobar hemorrhage (acute)
-
Cortical lesions (chronic)
-
Persistent paresis (50%)
Other risk factors include the following:
-
Language function deficit, dysarthria
-
Visual field defect (20%), hemianopia
-
Posture and balance deficit
-
Sensory, cognitive, and perceptual function deficits
-
Bowel and bladder incontinence
-
Deconditioning
-
Congestive heart failure
-
Hypertension
-
DM
-
Dysphasia
-
Spasticity
-
Contractures
-
Heterotopic calcification
Prognosis in Stroke
Significant improvement in UE function usually is seen only in the first 3 months poststroke. If no return of motor function is noted after more than 6 months, prognosis for useful function is unfavorable. If no return of voluntary motor function is noted after more than 1 week, it is unlikely that full use of the affected UE will return.
Poor prognostic indicators include the following:
-
Proprioceptive facilitation (tapping) response for more than 9 days
-
Traction response (shoulder flexors/adductors) in more than 13 days
-
Prolonged flaccid period
-
Onset of motion at longer than 2-4 weeks
-
Severe proximal spasticity
-
Absence of voluntary hand movement for more than 4-6 weeks
Stroke rehabilitation outcome
Predictors of outcome include the following:
-
Type, distribution, pattern, and severity of physical impairment [28]
-
Cognitive, language, and communication abilities
-
Number, types, and severity of comorbid conditions
-
Level of motivation or determination
-
Coping ability and coping style
-
Nature and degree of family and social supports
-
Type and quality of the specific training and adaptation program provided
Negative factors influencing the patient’s ability to return to work include the following:
-
Low score on Barthel index at the time of discharge from rehabilitation
-
Prolonged stay in rehabilitation
-
Aphasia
-
Prior alcohol abuse
Nonetheless, remarkable recoveries have been reported in 3-6 years, and patients have returned to work 3 years poststroke. [29]
A prospective study by Langhammer et al involving patients who, following a severe stroke, underwent specialized/comprehensive rehabilitation, concluded that a greater level of education, age less than 60 years, and a lesser amount of disability increases the likelihood that such patients will, by 6 months after discharge from the rehabilitation facility, return to work. The study included nine rehabilitation facilities in seven countries. [30]
Starting rehabilitation early correlates with better outcome but may be confounded by case severity. (See the graphs below.) However, stroke rehabilitation improves functional ability even in patients who are elderly or medically ill, as well as in those who have severe neurologic/functional deficits. Significant gains that are achieved are not attributable only to spontaneous recovery.
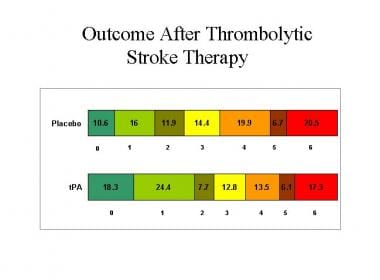 The bar graphs show the percentages of patients with stroke who demonstrated different outcomes on the modified Rankin Scale of global disability. These results were recorded 3 months following treatment of patients with tissue plasminogen activator (tPA) or placebo, in the National Institutes of Neurological Disorders and Stroke tPA trials 1 and 2. Rankin 0 = no symptoms; 1 = no significant disability, despite symptoms (able to perform all usual duties and activities); 2 = slight disability (unable to perform all previous activities but able to look after own affairs without assistance); 3 = moderate disability (requires some help, but able to walk without assistance); 4 = moderately severe disability (unable to walk without assistance and unable to attend to own bodily needs without assistance); 5 = severe disability (bedridden, incontinent, and requires constant nursing care and attention); 6 = dead. Image courtesy of UCLA Stroke Center.
The bar graphs show the percentages of patients with stroke who demonstrated different outcomes on the modified Rankin Scale of global disability. These results were recorded 3 months following treatment of patients with tissue plasminogen activator (tPA) or placebo, in the National Institutes of Neurological Disorders and Stroke tPA trials 1 and 2. Rankin 0 = no symptoms; 1 = no significant disability, despite symptoms (able to perform all usual duties and activities); 2 = slight disability (unable to perform all previous activities but able to look after own affairs without assistance); 3 = moderate disability (requires some help, but able to walk without assistance); 4 = moderately severe disability (unable to walk without assistance and unable to attend to own bodily needs without assistance); 5 = severe disability (bedridden, incontinent, and requires constant nursing care and attention); 6 = dead. Image courtesy of UCLA Stroke Center.
Of patients who survive stroke by more than 30 days, 10% demonstrate complete spontaneous recovery, 10% show no benefit from any treatment, and 80% may benefit from treatment. Stroke survivors who do not undergo rehabilitation are more likely to be institutionalized.
Eighty-five percent of patients went home after 3 months of participation in a stroke rehabilitation program. After 43 days, 80% of patients returned home, 85% were ambulatory, and 50-62% were independent in performance of ADL. Functional state improved in the stroke unit from 6-52 weeks.
Patients in outpatient and nonoutpatient therapy groups showed statistical improvement between stroke onset, discharge to home, and 1-year follow-up. The outpatient therapy group required a longer rehabilitation stay, was more impaired at onset, and did not perform as well as the nonoutpatient group. The outpatient therapy group was associated with complete UE/LE hemiplegia, unilateral neglect, impaired proprioception, and urinary incontinence.
Sphincter function, level of neurologic impairment, and capacity to perform ADL related to outcome are assessed, but these measures are not useful to anticipate the outcome of each patient.
Patients unable to walk 3 months poststroke received therapy up to 2 years after the stroke. Seventy-four percent of patients walked without assistance. Seventy-nine percent of patients had a modified Barthel score below 70.
Rehabilitation should include therapy directed at specific training of skills and functional training. Therapy should be given with sufficient intensity to promote skill acquisition.
A population-based study by Dhamoon et al suggests that within a group of patients who have suffered ischemic stroke, there will be an annual decline for up to 5 years in the proportion of patients who are functionally independent that is unrelated to recurrent stroke and other risk factors. [31]
In the study, 525 patients aged 40 years or older (mean age, 68.6 y) with incident ischemic stroke were prospectively followed at 6 months and annually for 5 years. During that time, the proportion of patients with a Barthel score of 95 or higher declined. The decline was independent of age, stroke severity, and other predictors of functional decline, occurring even in patients who did not suffer recurrent stroke or myocardial infarction. The authors also found that the decline occurred in patients who were receiving Medicaid or who had no health insurance but did not occur in those with Medicare or private insurance.
A study by Irisawa and Mizushima indicated that 4 weeks after admission to the stroke rehabilitation unit, poststroke functional recovery, as assessed using the motor Functional Independence Measure, is significantly linked to an absence of malnutrition (Geriatric Nutritional Risk Index score >92), a high body muscle percentage (>30% in males, >25% in females), and a high phase angle (>3.5° in males, >3.0° in females). [32]
Degree of recovery
The degree of recovery of independent functioning during rehabilitation has been found to be greater than that which might have been expected through a reduction in neural impairments alone, suggesting that rehabilitation interventions play an important role in the patient's recovery of function.
A retrospective study by Scrutinio et al involving 1265 patients with severe poststroke motor impairment found that patients’ motor scores, as measured using the Functional Independence Measure (FIM) rating scale, increased from a median 17 points on admission to a rehabilitation facility to a median 38 points at discharge. The investigators also found that at discharge, the severity of motor impairment had been reduced to a classification of mild (FIM-motor score of 62 or above) in 18.6% of patients, and moderate (FIM-motor score of 38-61) in 38.5% of them. [33]
The two types of poststroke improvement are related in subtle and complex ways. Alternative compensatory functional strategies, such as 1-handed dressing techniques for the hemiplegic patient, assume a major role in the performance of functional tasks when neurologic improvement is minimal or absent.
The degree of natural recovery of neurologic function varies, but figures on the relative frequencies of neurologic deficits during the early and later poststroke stages offer some insight into the degree of recovery that might be seen. The number of these deficits generally declines by approximately 33-50%. For example, the following reductions in prevalence from initial presentation have been found at 1-year follow-up:
-
Hemiparesis - From 73% at presentation to 37%
-
Aphasia - From 36% at presentation to 20%
-
Dysarthria - From 48% at presentation to 16%
-
Dysphagia - From 13% at presentation to 4%
-
Incontinence - From 29% at presentation to 9%
As previously mentioned, the time required for recovery also varies. Although most improvements in physical functioning occur within the first 3-6 months, later recovery also is commonly observed. Although it is tempting to specify a definitive prognosis in a stroke patient, it is important to recognize that a multiplicity of variables determine ultimate outcome, which is why expectations for recovery often are inaccurate.
Recovery and Diet
No special diet is required to improve the patient's motor recovery after stroke; however, the patient should avoid excessive weight gain and remain on the regular diet for conditions such as DM, hypertension, and hyperlipidemia.
A study did find evidence that intensive nutritional supplementation can improve motor recovery in previously undernourished patients who have suffered stroke and who are undergoing intensive inpatient rehabilitation. [34]
For excellent patient education resources, visit eMedicineHealth's Brain and Nervous System Center. Also, see eMedicineHealth's patient education article Stroke.
-
When the brain suffers an injury, such as a stroke, neurons release glutamate onto nearby neurons, which become excited and overloaded with calcium, after which they die (left). Normal neurotransmission (above) is altered during injury, causing excess calcium to activate enzymes, eventually leading to destruction of the cell. Since this process occurs via glutamate receptors, including N-Methyl-D-aspartate (NMDA) receptors, scientists believe that damage can be stopped through the use of agents that block these receptors.
-
Large intracerebral hemorrhage with midline shift.
-
The bar graphs show the percentages of patients with stroke who demonstrated different outcomes on the modified Rankin Scale of global disability. These results were recorded 3 months following treatment of patients with tissue plasminogen activator (tPA) or placebo, in the National Institutes of Neurological Disorders and Stroke tPA trials 1 and 2. Rankin 0 = no symptoms; 1 = no significant disability, despite symptoms (able to perform all usual duties and activities); 2 = slight disability (unable to perform all previous activities but able to look after own affairs without assistance); 3 = moderate disability (requires some help, but able to walk without assistance); 4 = moderately severe disability (unable to walk without assistance and unable to attend to own bodily needs without assistance); 5 = severe disability (bedridden, incontinent, and requires constant nursing care and attention); 6 = dead. Image courtesy of UCLA Stroke Center.
Tables

What would you like to print?
- Recovery Considerations
- Theories of Recovery
- Mechanisms of Recovery
- Pattern of Disability
- Pattern of Recovery
- PT Options in Stroke
- Occupational Therapy in Stroke
- Aphasia Therapy
- Associated Conditions
- Surgical Options
- Consultations in Stroke
- Other Treatment Options
- Home Care in Stroke
- Cardiac Precautions
- Complications During Rehabilitation
- Prognosis in Stroke
- Recovery and Diet
- Show All
- Media Gallery
- References





