Practice Essentials
Hereditary spastic paraplegia (HSP) is not a single disease entity; it is a group of clinically and genetically diverse disorders that share a primary feature, which is the causation of progressive and generally severe lower extremity weakness and spasticity. [1, 2, 3] (See Etiology, Presentation, and Workup.)
Strümpell first described hereditary forms of spastic paraplegia (see the image below) in 1883, with Lorrain later providing more extensive detail. [4] HSP is also called familial spastic paraparesis and Strümpell-Lorrain syndrome. (See Presentation.)
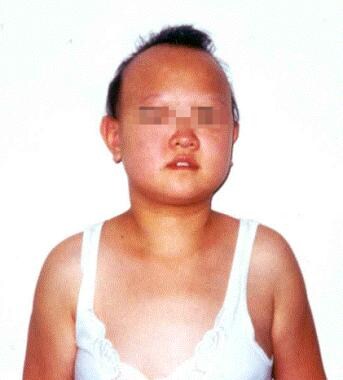 Photograph of a 16-year-old girl with complicated hereditary spastic paraplegia. She has a spastic gait disturbance, intellectual disability, and extrapyramidal symptoms. Note the dysmorphic features.
Photograph of a 16-year-old girl with complicated hereditary spastic paraplegia. She has a spastic gait disturbance, intellectual disability, and extrapyramidal symptoms. Note the dysmorphic features.
Numerous clinical reports have documented that HSP syndromes are heterogeneous. Syndromes are classified as uncomplicated, or pure, when only spinal involvement occurs, and they are classified as complicated when they are associated with neurologic abnormalities, such as ataxia, intellectual disability, dementia, extrapyramidal dysfunctions, visual or hearing dysfunctions, [5] adrenal insufficiency, and ichthyosis. Clinical distinctions between pure and complicated forms of HSP have some utility. (See Prognosis, Presentation, Workup, Treatment, and Medication.)
The most useful classifications now are based on the mode of inheritance and genetic linkage. HSP may also be classified as autosomal dominant, autosomal recessive, or X-linked, and each type has several subtypes, which are based on the location of the gene. The mode of inheritance cannot be used to predict the severity of the disorder, however, because symptoms can vary greatly within each type. (See Etiology.)
In the past, HSP was also classified as type I or type II, based on the patient's age at the onset of symptoms and on the amount of spasticity versus weakness. Because both types can appear in the same family, this method of classification is no longer in general use. (The age of onset often has no clear relation to the HSP genotype.) [6]
To date, the locations of several genes associated with HSP have been identified. Eighteen types of dominantly inherited pure or complicated HSP are known, along with 17 types of recessively inherited HSP and 3 types of X-linked HSP. [7]
Signs and symptoms of hereditary spastic paraplegia
In pure HSP, symptoms are generally limited to gradual weakening in the legs, urinary bladder disturbance, spasticity, abnormal gait, decreased sense of balance, and, sometimes, impaired sensation in the feet. [6]
In complicated HSP, a rare disorder, additional symptoms may include the following:
-
Peripheral neuropathy
-
Epilepsy
-
Ataxia
-
Optic neuropathy
-
Retinopathy
-
Dementia
-
Ichthyosis
-
Intellectual disability
-
Deafness
-
Problems with speech, swallowing, or breathing
Workup in hereditary spastic paraplegia
Genetic testing for SPG4/spastin mutations is available commercially, can provide laboratory confirmation of the diagnosis, and can be applied to prenatal testing.
Electrophysiologic studies are useful for assessing peripheral nerve, muscle, dorsal column, and corticospinal tract involvement in patients with HSP. [8]
Magnetic resonance imaging (MRI) scans may demonstrate atrophy of the spinal cords and occasionally of the cerebral cortex. [6] The cerebrospinal fluid in HSP is usually normal, although increased protein is noted in some patients.
Management
Currently, no specific treatment exists to prevent, retard, or reverse progressive disability in patients with HSP. Nonetheless, treatment approaches used for chronic paraplegia from other causes are useful.
Regular physical therapy (PT) is important for maintaining and improving range of motion (ROM) and muscle strength. Furthermore, PT is necessary to maintain aerobic conditioning of the cardiovascular system.
The types of exercise incorporated into PT programs for patients with HSP may include strengthening, stretching, and aerobic exercises.
Complications
Patients with HSP may have several possible complications, including the following (see Prognosis):
-
Gastrocnemius-soleus contracture
-
Cold feet
-
Fatigue
-
Back and knee pain
-
Stress and depression
A study by Schüle et al of 608 patients with HSP found that the ability to walk unassisted was maintained by these patients for a median 22 years’ disease duration but that independent walking ability was maintained longer by patients who had early onset disease. [9]
Etiology
HSP causes degeneration of the ends of the corticospinal tracts within the spinal cord. The ends of the longest fibers, which supply the lower extremities, are affected to a much greater extent than are the fibers to the upper body. Although some degeneration of the fibers supplying the arms commonly takes place, most people with HSP do not have symptoms in the hands or arms.
Impaired cellular membrane trafficking, more particularly, axonal transport of macromolecules and organelles, is the best-characterized genetic mechanism of HSP. Several proteins, such as spastin (SPG4) and atlastin-1 (SPG3A), which shape membranes of the endoplasmic reticulum or endosomes, are known as such candidates. [10, 11]
Mitochondrial dysfunction is the second process that leads to HSPs. Paraplegin (SPG7) is a candidate for the development of such dysfunction. It is part of the m-AAA protease, an adenosine triphosphate (ATP) ̶ dependent proteolytic complex located at the mitochondrial inner membrane, which controls protein quality and regulates ribosome assembly. [12, 13]
In most cases of HSP, the primary problem may be disturbance of the ends of the long axons, with little or no loss of myelin and no abnormal myelin. A rare type of X-linked HSP, however, has been associated with a myelin protein gene mutation. Patients with this form of HSP generally show evidence of myelin abnormalities, which are known to affect axon function. Although genes involved with myelination of the central nervous system (CNS) are less likely to be involved with HSP than are those associated with axonal stability, these genes must be considered.
A study by Agosta et al suggested that the various neurologic disorders designated as HSP share a common neurodegenerative cascade. Magnetic resonance imaging (MRI) revealed that in patients with different clinical pictures, a similar involvement existed for the motor, association, and cerebellar white matter pathways and for the cervical cord, in relation to healthy controls. [14]
Genetics
Presently, more than 80 genetic loci have been identified. There are families with autosomal dominant patients and with autosomal recessive and sporadic patients. In a report on HSP in Japan, Koh et al stated that causative genes could not be found in 35% of autosomal dominant patients or in 52% of autosomal recessive and sporadic patients. [7, 15, 16, 17, 18, 19, 20, 21, 22]
Most cases of pure HSP are autosomal dominant, whereas complicated forms tend to be autosomal recessive. With regard to pure, autosomal dominant HSP, SPG4, SPG3A, and SPG6 account for 70-80% of families. [13]
SPG4 HSP is the single most common dominantly inherited HSP, representing approximately 40% of such cases. Hazan and colleagues discovered that mutations in a novel gene designated SPG4 (protein, spastin) are the cause of this disorder. [23] Insights into the SPG4 phenotype and spastin function can yield useful information relating to hypotheses for axonal degeneration in SPG4 HSP, such as direct cytoskeletal instability, abnormal mitochondrial distribution, and other consequences of abnormal axonal transport. [16, 13, 24, 25]
A second autosomal dominant HSP (SPG3A) shows a linkage to band 14q11-q21 and accounts for approximately 10% of cases. This is also a pure HSP. Symptoms usually begin in early childhood and are often nonprogressive. Genetic testing for SPG3A is commercially available.
A third autosomal dominant HSP, SPG6, shows a linkage to band 15q11.1. Symptoms begin in late teenage years. This kindred contains a number of affected members who have developed more severe disability than typical HSP families with other linkages. Penetrance is age dependent and high. Other genes involved in autosomal dominant HSPs are SPG8, SPG10, SPG13, SPG31, and SPG33.
SPG5, SPG7, and SPG11 are involved in autosomal recessive HSPs. A family with pure HSP demonstrated a linkage to band 8q12-q13 (SPG5 HSP). SPG7 HSP has been linked to mutations in the gene encoding for paraplegin and accounts for around 5% of autosomal recessive HSPs. [16, 26] This type of mutation produces both pure and complicated HSP phenotypes. Mutations in the gene result in impaired oxidative phosphorylation.
SPG11 HSP, which is characterized by a thin corpus callosum, is a clinically distinct form that includes cognitive impairment and severe axonal neuropathies. [18, 27, 28] A study by Faber et al indicated that in SPG11 HSP, selective neuronal vulnerability exists, with white matter involvement being precocious and widespread and subsequent gray matter degeneration being restricted but progressive. [29]
X-linked HSP is complex but rare, and the border between pure and complicated HSP syndromes is blurred. SPG1 HSP is linked to mutations in the gene for the L1 cell adhesion molecule (L1CAM); these mutations are associated with hydrocephalus, spasticity, ataxia, intellectual disability, and adducted thumbs.
SPG2 HSP is linked to a duplication mutation in the gene for proteolipid protein, which is located on band Xq21-q22. Mutations in this gene are also related to complicated X-linked HSP and to the dysmyelinating condition Pelizaeus-Merzbacher syndrome. One other rare X-linked form of HSP has been described (associated with SPG16). Affected individuals have quadriplegia, motor aphasia, reduced vision, mild intellectual disability, and sphincter disturbance. [30]
Preliminary genotype-phenotype correlations
With the identification of HSP loci on chromosome X and 2p, 8q, 14q, 15q, and 16q, a comparison of phenotypes is possible in families for whom the disorder is linked to one of these loci, as well as in HSP families for whom these loci are excluded. [31]
Thus far, genetically diverse types of autosomal dominant HSP (those linked to 2p, 14q, and 15q) appear to be clinically and electrophysiologically similar. This observation suggests that the different abnormal gene products may interact in a common biochemical cascade that results in similar patterns of neuronal degeneration.
The disorder may be more severe in the 15q-linked kindred than in kindreds linked with 14q. In a study of the kindred with disease linked to 14q, only 1 patient needed a wheelchair. [32] In contrast, 9 of the patients affected in a kindred HSP linked to 15q required a wheelchair (for some patients, the need began in their 40s).
Kindreds with autosomal dominant HSP linked to 2p have exhibited (1) the prototypical adolescent- or adult-onset, progressive form and (2) the less common childhood-onset, relatively nonprogressive form. The significant variations in patients' ages at symptom onset and the degree of progression in these kindreds indicate that the complete phenotype is influenced by different mutations in the same gene or by the effects of modifying genes.
Epidemiology
In Europe, the frequency of HSP is estimated to be 1-9 cases per 100,000 population. A study by Ortega Suero et al estimated that the prevalence of HSP in Spain is 2.24 cases per 100,000 population. [33] Because HSP is rare, it is often misdiagnosed, making the actual frequency difficult to determine. A reasonable estimate, however, is that it affects approximately 3 persons per 100,000 population. This represents fewer than 10,000 cases in the United States. Further estimates indicate that about 10% of people with HSP have complicated HSP.
Pure HSP may occur at any age, from infancy through late adulthood. However, most patients experience the onset of symptoms between the second and fourth decades of life.
Prognosis
In patients with pure HSP, life expectancy typically is unaffected by the condition. Generalizations about the life expectancy of people with complicated HSP are difficult to make, because each patient has unique symptoms. Possible complications associated with HSP are described below.
Gastrocnemius-soleus contracture
This condition is more common when symptoms begin in childhood. It also occurs when physical therapy (PT) has not been sufficient.
Cold feet
Many people with HSP complain of cold feet. This is a common complaint in many disorders of the upper or lower motor neurons. Cold feet may be related to abnormal thermoregulation of cutaneous vessels; however, circulation is usually preserved.
Fatigue
Fatigue is a common symptom of HSP. One obvious cause is the extra effort required for walking, because of muscle weakness in the legs. In addition, various medications prescribed for HSP cause drowsiness or fatigue. Moreover, many patients with HSP may not get the amount of sleep they require, because of leg cramps or spasms or as a result of the frequent need to urinate during the night. A less obvious cause of fatigue may be the fact that, because of a more sedentary lifestyle, people with HSP often are not aerobically fit and therefore have reduced endurance. Stress or depression also can contribute to fatigue.
Back or knee pain
Back and/or knee pain is common in people with HSP. The pain is not directly due to HSP itself but is instead often caused by muscle weakness and gait abnormalities resulting from HSP. As certain muscles become weaker, other muscles need to compensate for that weakness. Compensatory measures create an awkward gait that causes strain on many muscles and joints. Patients may thrust their shoulders back or swing their legs outward as they walk. Use of certain mobility devices also may put a strain on the arms or back.
Stress and depression
Stress, depression, and denial are not unusual in patients with HSP or any other chronic illness. Denial is not necessarily a problem, as long as the person in denial is not depriving himself or herself of proper treatment and care. Denial allows some people to cope and set their worries aside. However, some people with HSP face denial by family members who refuse to admit that a problem exists. This can create a frustrating and stressful situation.
-
Photograph of a 16-year-old girl with complicated hereditary spastic paraplegia. She has a spastic gait disturbance, intellectual disability, and extrapyramidal symptoms. Note the dysmorphic features.
-
Dysmorphic appearance of a 16-year-old girl with complicated hereditary spastic paraplegia. This patient displays a short stature (145 cm) and hair loss. Anterior (left), lateral (middle), and posterior (right) views are shown.
-
General appearance of sisters with complicated hereditary spastic paraplegia. They are aged 16 and 17 years. Physical examination revealed increased deep tendon reflexes in all 4 extremities, with an extensor plantar reflex. Sensory losses in the patients have affected mainly their joint positions and vibration sensations.







