Overview
Chronic wounds represent a significant burden to patients, health care professionals, and the US health care system, affecting 5.7 million patients and costing an estimated $20 billion annually. [1, 2] To effectively manage these problems, one must understand the normal healing process and engineer a salubrious physical and biochemical environment. This article outlines normal healing biology, describes the factors that facilitate or impair wound healing, surveys common types of problem wounds, and discusses emerging concepts in chronic wound management.
For more information on wound management, including news and CME, visit Medscape’s Wound Management Resource Center.
The Biology of Wound Healing
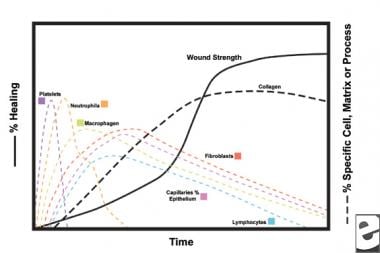
With the wounding of healthy tissue, a predictable progression of physiologic events unfolds. This progression can be divided into the phases of inflammation, proliferation, and maturation. Each phase is characterized by the sequential elaboration of distinctive cytokines by specific cells. See the images below.
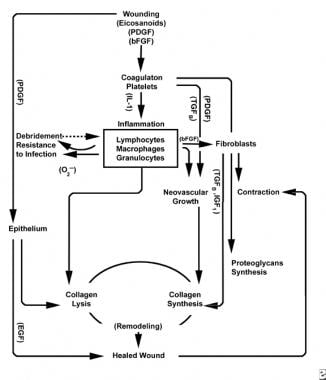
The inflammatory phase
The inflammatory phase simultaneously launches hemostatic mechanisms and pathways that create the clinically recognizable cardinal signs of inflammation: rubor (redness), calor (warmth), tumor (swelling), dolor (pain), and functio laesa (loss of function).
Injury to vascular tissue initiates the extrinsic coagulation cascade by releasing intracellular calcium and tissue factor that activate factor VII. The resulting fibrin plug achieves hemostasis aided by reflex vasoconstriction. This plug acts as a lattice for the aggregation of platelets, the most common and “signature” cell type of the early inflammatory phase.
Platelets elaborate a number of proinflammatory substances, such as adenosine diphosphate, tissue growth factor beta (TGF-ß), and platelet-derived growth factors (PDGF). These growth factors act on surrounding cells and stimulate chemotaxis of neutrophils, monocytes, and fibroblasts to the area of injury. [3]
Injured tissues, through activated phospholipase A, simultaneously catalyze arachidonic acids to produce vasoactive prostaglandins and thromboxane, collectively known as eicosanoids. Eicosanoids mediate activity influencing platelet plug formation, vascular permeability, and cellular chemotaxis to influence wound healing. For example, thromboxane A2 mediates vasoconstriction and platelet aggregation. [3]
After initial vasoconstriction, the classic signs of inflammation manifest from increased vascular permeability. Rubor results from vasodilation, mediated by prostacyclin (PGI2), prostaglandin A (PGA), prostaglandin D (PGD), and prostaglandin E (PGE). Tumor and calor develop as vascular endothelial gaps enlarge, allowing the egress of plasma protein and fluid into the interstitial space. These changes are potentiated by PGE2 and prostaglandin F2α (PGF2α) and allow the ingress of inflammatory cells into the area of injury, including cells that elaborate. Dolor is sensed as PGI2, PGE, and PGE2 act on peripheral nociceptors. [4]
In the second stage of the inflammatory phase, leukocytes supplant platelets as the dominant cell type, attracted by chemotaxis. White blood cells (WBCs) are the predominant cells for the first 3 days after wounding; their numbers peak at approximately 48 hours. Polymorphonucleocytes (PMNs) are the first to begin bactericidal activities using inflammatory mediators and oxygen free radical metabolites. However, normal wound healing can occur without PMNs. Another leukocyte, the helper T cell, elaborates interleukin-2 (IL–2). IL-2 promotes further T cell proliferation to augment the immunogenic response to injury.
As PMN leukocytes begin to wane after 24-36 hours, circulating monocytes enter the wound and mature into tissue macrophages. These cells debride the wound on the microscopic level and produce a wide variety of important substances, such as IL-1 and basic fibroblast growth factor (bFGF). IL-1 stimulates the proliferation of inflammatory cells and promotes angiogenesis through endothelial cell replication. bFGF is a chemotactic and mitogenic factor for fibroblasts and endothelial cells. Unlike PMNs, macrophage depletion severely impairs wound healing, as debridement, fibroblast proliferation, and angiogenesis all diminish.
Toward the end of the inflammatory cycle, the evolving milieu of eicosanoids in the wound interact with the cell types present, resulting in fibroblast synthesis of collagen and ground substance (from increased ratio of PGF2α to PGE2). Additionally, the macrophage-derived growth factors are now at optimal levels, strongly influencing the influx of fibroblasts and then keratinocytes and endothelial cells into the wound. As mononuclear cells continue to replace WBCs and macrophages, the proliferative phase begins.
The proliferative phase
Two to three days after wounding, fibroblasts migrate inward from wound margins over the fibrinous matrix established during the inflammatory phase. During the first week, fibroblasts begin producing glycosaminoglycans and proteoglycans, the ground substance for granulation tissue, as well as collagen, in response to macrophage-synthesized bFGF and TGF-ß, as well as PDGF.
Fibroblasts soon become the dominant cell type, peaking at 1-2 weeks. They generate not only collagen molecules but also cytokines such as PDGF, TGF-ß , bFGF, keratinocyte growth factor, and insulinlike growth factor-1. Fibroblasts also assemble collagen molecules into fibers, which are cross-linked and organized into bundles. Collagen is the major component of acute wound connective tissue, with net production continuing for the next 6 weeks. The increasing content of wound collagen correlates with increasing tensile strength. [5, 6]
Keratinocytes and endothelial cells also proliferate during this time, eventually producing autocrine growth factors that maintain their growth. Endothelial expansion contributes to angiogenesis, as intact vessels generate buds in granulation tissue. Neovascularization facilitates growth of the advancing line of fibroblasts into the wound, providing them with necessary nutrients and cytokines.
Degradation of the fibrin clot and provisional matrix is accompanied by the deposition of granulation tissue (ground substance, collagen, capillaries), which continues until the wound is covered. Decreasing hyaluronic acid (in ground substance) levels and increasing chondroitin sulfate levels slow fibroblast migration and proliferation while inducing fibroblast differentiation, transitioning to the maturation phase of wound healing.
The maturation phase
For the first 6 weeks, new collagen production dominates the wound healing process, deposited randomly in acute wound granulation tissue. As the wound matures, collagen is remodeled into a more organized structure with increased tensile strength. Gradually, type I collagen replaces type III until the normal skin ratio of 4:1 is achieved. As remodeling continues, matrix metalloproteinase collagenolysis achieves a steady state with collagen synthesis. Tensile strength plateaus at 80% of the original strength approximately 1 year postinjury. [7, 8, 9]
Superficial to this activity, epithelial cells continue to migrate inward from the wound edge until the defect is covered. At this point, contact inhibition induces transformation of fibroblasts into myofibroblasts, which contain contractile actin fibers. Wound contraction follows, replacing injured tissue volume with new tissue, although the exact role of the myofibroblast has not been fully elucidated. [4]
Deterrents to wound healing
Acute wounds generally proceed through an orderly and timely reparative process that results in a durable restoration of anatomic and functional integrity. However, various physiologic and mechanical factors may impair the healing response, resulting in a chronic wound that fails to proceed through the usual stepwise progression. Local infection, hypoxia, trauma, foreign bodies, or systemic problems such as diabetes mellitus, malnutrition, immunodeficiency, or medications are most frequently responsible.
All wounds are contaminated, but most successfully resist invasive infection. When the concentration exceeds 100,000 (105) organisms per gram of tissue or the immune system becomes compromised, infection frequently ensues. [10] Cellulitis prolongs the inflammatory phase by maintaining high levels of proinflammatory cytokines and tissue proteases, which degrade granulation tissue and tissue growth factors, and by delaying collagen deposition. [11, 12]
Debridement (surgical, enzymatic, and/or by dressing changes) and antibiotics are the mainstays of antibiotic treatment. Debridement removes devitalized tissue, which can be a source of endotoxins that inhibit fibroblast and keratinocyte migration into the wound. Foreign bodies may also require removal, as the presence of a silk suture reduces the number of bacteria required to incite infection 10,000-fold. [12] (For a detailed description of technique, see Medscape Reference article Wound Foreign Body Removal.)
Cellular hypoxia retards wound healing through various means. Collagen fibril crosslinking requires oxygen to hydroxylate proline and lysine and fails when tissue pressure is below 40 mm Hg. [13] The bactericidal potency of leukocyte oxidative phosphorylation also suffers in a hypoxic environment, reducing the threshold for infection. Measures to improve oxygen delivery depend on the etiology. Tobacco use, which causes vasoconstriction and increases platelet adherence, should be stopped. Angioplasty or arterial bypass grafting may be required for peripheral vascular disease. Adjunctive measures to improve systemic perfusion in cases of cardiac failure may be indicated. Hematocrit value less than 15% should be treated and euvolemia restored, as needed. Venous stasis or lymphatic insufficiency may be improved with compressive garments.
Systemic disease can dramatically prolong or interrupt wound healing. Glycosylation in diabetes mellitus impairs neutrophil and macrophage phagocytosis of bacteria, prolonging the inflammatory phase. The proliferative phase is also protracted in the same disease as erythrocytes become less pliable and less able to deliver oxygen to the wound for tissue metabolism and collagen synthesis. [14]
In a multicenter study of 1000 patients with chronic leg ulcers, Jockenhöfer et al found that comorbidities in these individuals included arterial hypertension (70.5%), obesity (45.2%), non-insulin dependent diabetes (27.2%), dyslipidemia (24.4%), and metabolic syndrome (18.4%). [15]
Malnutrition results in diminished fibroblast proliferation, impaired neovascularization, and decreased cellular and humoral immunity. Wounds exert heightened metabolic demands, particularly within granulation tissue. Amino acids such as methionine, proline, glycine, and lysine, are essential for normal cell function and the repair of cutaneous wounds. Fatty acids are critical constituents of cell membranes and are the substrate for the eicosanoids that mediate the inflammatory process. Essential fatty acids linolenic and linoleic acid must be supplied in the diet, as the human body is incapable of de novo synthesis of these molecules. [16]
Adequate vitamins and minerals must be available for cell metabolism, acting as cellular signals and cofactors. Vitamin C (ascorbic acid) and iron are required for the hydroxylation of lysine and proline, which crosslink and stabilize the triple helix structure of collagen; copper also plays an role in stabilizing collagen. Vitamin A (retinoic acid) plays an important role in modulating collagen production and degradation and is particularly important in epithelialization. A potent antioxidant, vitamin E (alpha tocopherol) appears to accelerate dermal and bone healing in animals, and supplementation may have a role in humans. Trace metal, particularly zinc, deficiency is also associated with poor wound healing; this should be replenished, as appropriate. [17]
Ovid purportedly wrote, “medications sometimes heal, sometimes kill.” This is certainly true regarding wound healing. Corticosteroids blunt the processes of the entire inflammatory phase. Vitamin A (topically or 25,000 IU/d orally) mitigates the detrimental healing effects of corticosteroids, but hepatotoxicity may result from prolonged use (ie, >1 mo). Nonsteroidal antiinflammatory medications (NSAIDs) also interfere with arachidonic acid metabolism and, therefore, wound healing. Additionally, NSAIDs inhibit platelet function, one of the earliest processes in the inflammatory phase. [18]
A study by Sutcliffe et al suggested that upregulation of the gap junction protein connexin is common to chronic wounds. Examining connexin in three types of wounds—venous leg, diabetic foot, and pressure ulcers—the investigators found that each type of wound displayed upregulation of epidermal connexin 43, connexin 26, and connexin 30, as well as dermal connexin 43. [19]
Common Chronic Wounds
Common chronic skin and soft tissue wounds include the diabetic foot ulcer, the pressure ulcer, and the venous stasis ulcer. [20]
Nonetheless, a retrospective study by Khoobyari et al of 270 chronic ulcer biopsy specimens found that 44.8% of the biopsied ulcers arose from atypical causes, most commonly neoplasms (basal cell carcinoma, squamous cell carcinoma, melanoma, cutaneous T-cell lymphoma). The investigators concluded, therefore, that although routine histology by itself cannot determine the etiology of many ulcers, skin biopsy is still an essential workup tool for chronic ulcers considering the significant proportion of these lesions that develop via atypical causes. [21]
Diabetic foot ulcer
Diabetic ulcers are responsible for most foot and leg amputations in the United States. Patients with diabetes develop these ulcers with an incidence of 2% per year; a study by Rice et al indicated that for Medicare and private insurers, diabetic foot ulcers add $9-13 billion in additional diabetes costs. [22, 23, 24, 25, 26] Pathogenesis of the ulcers is due to neuropathic impairment of musculoskeletal balance, as well as immune compromise from leukocyte dysfunction and peripheral vascular disease, complicating these wounds with infection. [14] Standard of care includes off-loading, attentive debridement, maintenance of a moist wound environment, and, when cellulitis is present, systemic antibiotics.
Chronic wounds have decreased levels of growth factors, and topical platelet-derived growth factor (PDGF), tissue growth factor beta (TGF- ß), and platelet-derived wound healing factor have been demonstrated to speed the healing of diabetic ulcers. The PDGF becaplermin (Regranex gel, Johnson & Johnson) was the first recombinant growth factor to be approved by the US Food and Drug Administration for such use.
A study by Chen et al indicated that following hospital treatment for diabetic foot ulcer, invasive systemic infection associated with the ulcer (DFU-ISI) is an important late complication that increases mortality risk. In the study’s patients, methicillin-resistant Staphylococcus aureus (MRSA) gave rise to 57% of the ISIs. Using Cox regression modeling, the investigators found that complicated ulcer healing and the presence of MRSA in the initial ulcer culture predicted the development of DFU-ISIs, with the hazard ratio for mortality in association with DFU-ISIs being 1.987. [27]
Pressure ulcer
Pressure ulcers result from ischemia due to prolonged pressure over a bony prominence. They typically occur in paralyzed or unconscious patients who unable to either sense or respond to the need for periodic repositioning. [28]
A study by Cowan et al found that in patients with paralysis, the rate of pressure ulcer development differed according to the type of paralysis suffered. In patients with paraplegia, quadriplegia, or hemiplegia, the prevalence of pressure ulcers (stage 2, 3, and 4 lesions; unstageable pressure ulcers; and lesions with suspected deep-tissue injury) was 47.4%, 33.9%, and 9.6%, respectively. [29]
A literature review by Shiferaw et al indicated that globally, there is a 32.36% pooled prevalence of pressure ulcers in patients with spinal cord injury. This suggested that pressure ulcers occur much more frequently in association with spinal cord injury than among patients in general, with a previous study having found the global prevalence of pressure ulcers in public hospitals to be 14.8%. [30]
Preventive measures against pressure ulcers include identification of high-risk patients, frequent assessment, scheduled repositioning, pressure-relief bedding, moisture barriers, and adequate nutritional status. [31] Treatment consists of pressure relief, enzymatic and surgical debridement, and maintenance of a clean, moist wound environment. [32, 33, 34, 35]
Topical antibiotics and PDGF may have some role in treatment, while the Vacuum-Assisted Closure Device (Kinetic Concepts, Inc, San Antonio, Tex), has definitively demonstrated significant benefits in accelerating wound healing. [36, 37, 38] Ultimately, many wounds require ostectomies and flap coverage for definitive management. (Click here to complete a Medscape CE activity on pressure ulcers.)
Venous stasis ulcer
Venous stasis ulcers result from hypoxia in areas of venous congestion in the lower extremity. Possibly, the thick perivascular fibrin cuffs impede oxygen diffusion into surrounding tissues. Alternately, macromolecules leaking into perivascular tissue trap may growth factors needed for the maintenance of skin integrity. A third potential cause may be leukocytes migrating through capillaries more slowly than usual, even occluding them, becoming activated, and damaging the vascular endothelium. [39]
Compression hose or boots, debridement, and maintenance of a clean, moist wound environment are the mainstays of therapy. Split-thickness skin grafts and bioengineered skin equivalent (Apligraf, Organogenesis) have both been shown to be effective, providing matrices, migration pathways, growth factors, and living dermal and epidermal cells to the wound. [40, 41] In addition to compressive bandaging, surgery to correct venous reflux does not appear to improve ulcer healing, though it may reduce the recurrence of problem wounds. [37]
Emerging Trends in Wound Healing
Hyperbaric oxygen significantly increases the oxygen saturation of plasma, raising the partial pressure available to tissues. Recent Cochrane data analysis concluded that hyperbaric therapy for diabetic foot ulcers can significantly reduce the risk of major amputation, but that for other chronic wounds, the routine application of this therapy is not currently justified. [13]
Adjuncts to surgical wound debridement can help remove excess fluid from the wound, hastening healing. Negative pressure wound therapy has revolutionized wound care in both civilian and military wounds. [42, 43, 44, 45] Nonmechanical topical products such as cadexomer, which absorbs up to 6 times its own weight in exudate and transudate, serve a similar purpose and have been shown to help accelerate wound closure in chronic lesions. [46]
Biologically-derived molecules from humans as well as other species may find more clinical applications in the future. Dirhamnolipids are a class of molecules produced by Pseudomonas species that, when applied topically, have been reported to improve healing in chronic wounds in mice and humans. [47] Cell adhesion proteins such as amelogenin, when topically added to compression therapy, appear to promote ulcer healing in the lower extremity. [48]
Therapies directed at autologous cytokines and enzymes may also prove increasingly fruitful. Metalloproteinases such as ADAM 12 appear to play a regulatory role involving growth factors and are increased in chronic ulcers, leading some to speculate that therapy directed at ADAM 12 may prove useful. [49] Other, systemic molecules, such as angiotensin II, have recently been described as acting on keratinocyte and fibroblast migration through a heparin-binding epidermal growth factor (EGF)-like growth factor, providing another potential future target for intervention. [50, 51, 52]
Perhaps most intriguing, recent developments in mesenchymal stem cell therapy have been applied to problem wounds with some success, providing injured tissue with pluripotential cells that develop into durable tissue and elaborate growth factors and cytokines. [53, 54] Yoshikawa et al demonstrated wound healing in previously refractory wounds with autologous marrow mesenchymal stem cells implanted in a collagen dermal substitute for human burn, pressure, and venous ulcers. [55, 56, 57]
A study by Eschborn et al suggested that in chronic wounds being healed through secondary intention, the use of adipose-derived stem cells, delivered through autologous fat transfer, can aid in wound reduction and closure. [58]
Therapy with genes encoding for growth factor and/or cytokines used independently or with stem cells may also eventually prove to be useful in treating wounds resistant to more traditional approaches; Branski et al provide a lucid outline of these technologies. [1]
-
Schemes of the wound healing process.
-
Cellular characteristics of the wound healing process.








