Overview
The growth of the human calvaria is a complex process that is not yet completely understood. It begins with growth from 5 primary ossification centers meeting to form 6 main suture sites. Undisturbed, this process culminates in a normally shaped adult head. Craniosynostosis refers to a specific aberration of the growth process in which bone growth at a particular suture site (or sites) is arrested prematurely, resulting in a specific skull shape deformity. This article discusses the development of the skull; pathogenesis, biology, and genetics of craniosynostosis; and its surgical treatment. [1, 2]
Embryology and Development
The human cranium develops from mesoderm and neural crest cells. Beginning at 5 weeks, the mesenchymal tissue organizes to form cartilaginous plates and later, bone that comprises the skull. The neurocranium (calvaria in the adult) and viscerocranium (facial bones and portions of cranial base in the adult) combine to form the skull and grow independently through separate mechanisms.
The neurocranium develops in two distinct sections, cartilaginous and membranous. The cartilaginous neurocranium (or chondrocranium) comprises a number of cartilaginous plates that grow, ossify, and fuse to form most of the cranial base. Parachordal cartilage, located at the end of the notochord, combines with occipital somites to form the base of the occipital bone, later growing to boundaries of the foramen magnum. Slightly more rostral, the hypophyseal cartilages coalesce to form the body of the adult sphenoid bone. Trabeculae cranii fuse to form the body of the adult ethmoid bone. Nasal capsules form the rest of the ethmoid bone.
Laterally, the alae orbitalis grows to become the lesser wing of the sphenoid bone and the alae temporalis grows to become the greater wing. The otic capsules develop into the petrous portion of the temporal bone and the mastoid process. These portions of the skull undergo endochondral ossification and form the greater portion of the cranial base, contributing little to the cranial suture involved in most syndromal and nonsyndromal craniosynostoses.
The membranous neurocranium is derived from mesenchyme that invests the developing brain, a relationship that may help explain the etiology of diseases of premature suture fusion. Through membranous ossification, bone spicules form in 5 primary ossification centers, paired frontal and parietal centers, and a single occipital center. Growth at these ossification centers proceeds in a radial fashion with new bone formation (osteoblastic) at the edges and osteoclastic resorption toward the center. The flexible membranous junctions between neurocranial bones, termed sutures, are formed where growth from two ossification centers meet. In areas where more than two ossification centers join, a fontanelle forms.
The presence of sutures in the cranial skeleton allows for compression and overriding of cranial bones during birth. Often distortion of the head persists for several days or weeks after birth. In most patients this deformity corrects spontaneously during rapid postnatal growth. Though this purpose for cranial sutures seems teleologically sound, Murray observes that cranial sutures are present in the skulls of chickens who do not suffer the forces of passage through a birth canal. While the molding function of sutures may be questioned, normal growth relies on their patency and appropriately timed closure.
The 6 fontanelles present at birth are the anterior, posterior, and paired anterolateral and posterolateral fontanelles. The posterior fontanelle closes first at about 2 months. The anterolateral fontanelles (corresponding to the pterion in the adult) normally close 3 months after birth. The posterolateral fontanelles (corresponding to the asterion in the adult) close at the end of the first year, and the anterior fontanelle, which is the last to close, generally does so by 2 years.
The mendosal suture closes first, several days after birth. The mendosal is an obscure suture in the occipital bone running horizontally from the medial portion of the lambdoid suture. The median frontal, or metopic, suture usually closes by 2-6 years. In 10% of the population it remains open until adulthood, and the frontal sinuses are absent or hypoplastic. The remaining sutures close clinically at 6-12 months but do not ossify completely until after 30 years.
The neurocranium achieves 63% of its ultimate growth by birth, 88% by 1 year, and 95% by 10 years. The neonatal brain doubles in volume by 6 months and triples in volume by 2.5 years. By 16 years, the calvaria achieves its mature size and only changes slightly as the bone thickens and remodels over the following 3-4 years.
Although originally considered the primary center of growth, sutures currently are believed to grow in response to expansion of soft tissues, specifically the brain, and more directly, the dura. The proximity of the dura to the suture is critical in maintaining patency. If a strip craniectomy is performed, the defect will heal and fuse again if no effort is made to restrict this growth. Babler demonstrated that changes in growth of a particular suture affect growth patterns of neighboring sutures. [3, 4] The exact mechanism of sutural growth and closure is not yet fully understood, but researchers continue to investigate the molecular biology controlling this process, which is discussed later in this article.
The image below shows normal cranial suture anatomy at birth.
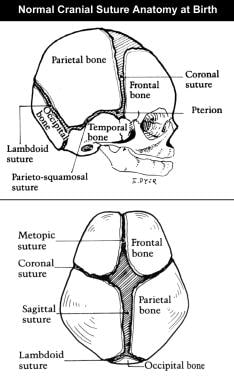
Pathogenesis
Söömmering published the first discussion of craniosynostosis in modern literature in 1791. He hypothesized that cranial deformities were caused by a failure of growth along cranial sutures. Otto echoed this notion in 1831 and observed craniosynostosis secondary to microcephaly. The modern understanding of sutural growth and synostosis is rooted in the writing of Virchow, who described the pattern of calvarial growth. [5]
According to Virchow's law, cranial growth takes place at right angles to the direction of the sutures, thus premature ossification of a suture results in growth arrest in a direction perpendicular to the suture. Compensatory growth is not explained fully by Virchow's law except that it implies that such growth may occur in the direction of the suture. Delashaw further describes compensatory growth, listing 4 fundamental factors that explain compensatory growth patterns. [6]
Cranial vault bones that are prematurely fused act as a single bone plate with decreased growth potential.
Abnormal asymmetric bone deposition occurs at perimeter sutures with increased bone deposition directed away from the bone plate.
Perimeter sutures adjacent to the prematurely fused suture compensate in growth more than perimeter sutures distant to the sutural stenosis.
A nonperimeter suture that is contiguous to the prematurely fused suture undergoes enhanced symmetric bone deposition along both edges.
The exact cause of premature fusion of sutures is not understood fully. Researchers have proposed 3 theories explaining the etiology of craniosynostosis. Virchow submitted that fusion of a cranial suture is a primary event causing a secondary deformation of the cranial base. Conversely, Moss hypothesizes that cranial base malformation is a primary event resulting in deformity of the cranial vault. [7] Finally, Park and Powers suggest that a defect of the mesenchymal blastema causes malformation of both the cranial vault and cranial base.
Primary and secondary causes
Craniosynostosis may be primary, occurring without an identifiable causative agent, or secondary. In most patients, craniosynostosis is a primary event and occurs sporadically. While the mechanism of primary craniosynostosis remains to be elucidated, there are many known secondary causes.
Craniosynostosis may be induced through internal and external forces. Environmental (external) forces can cause premature fusion of sutures. Prenatal compression of the fetal head is implicated as a cause of craniosynostosis. Similarly, Persson described a method for inducing immobilized coronal sutures in rabbits with application of methyl 2-cyanoacrylate adhesive. This reliably resulted in fusion of the treated suture, although this was performed postnatally.
Persson theorized that sutures fused from mechanical restriction caused by rigid bridging with acrylic cement. These results fell under scrutiny when Nappen and Kokich attempted to isolate the suture from the toxic effects of the adhesive by placing a sheet of polyethylene film over the suture site. Despite the same rigid restriction of the suture, no synostosis occurred. However, Koskinen-Moffett and Moffett, who clipped the cervices of pregnant mice causing a delay in birth, confirmed prenatal restrictive forces as a cause for craniosynostosis. This manipulation led to an 88% rate of craniosynostosis of squamosal sutures. However, this suture is rarely prematurely fused clinically, and this finding may represent an anomaly of the animal model chosen.
Internal forces known to cause craniosynostosis include a number of teratogens and diseases. Teratogens implicated in craniosynostosis include aminopterin, diphenylhydantoin, valproic acid, oxymetazoline hydrochloride, and possibly isotretinoin. Craniosynostosis has been observed in association with a number of maternal metabolic disorders including hyperthyroidism, rickets, Hurler syndrome, Morquio syndrome, beta-glucuronidase deficiency, mucolipidosis III, and a host of hematologic disorders.
Internal forces also may be mechanical. In shunted hydrocephaly, for example, the normal expansion of cranial contents is abruptly halted. The resultant lack of sutural stretch is associated with synostosis. In microcephaly, the lack of brain growth leads to an absence of normal stretch stimulus across the suture. Cranial sutures may be poorly defined or absent.
As previously discussed, the dura plays an important role in maintaining sutural patency and is a likely contributor to premature closure. In microcephaly, abnormal or incomplete dura may be partially responsible for the limited growth of the cranial vault and the abnormal absence of sutures. Sporadic absence of the metopic suture in holoprosencephaly supports this idea. Variable anatomy in each patient may lead to an inconsistent relationship of dura to the area of the metopic suture, resulting in formation of this suture only in patients with good apposition of dura to the frontal bone.
Recent work of Levine et al further supports the importance of the dura to sutural function. [8] Levine et al performed strip craniectomies across two cranial sutures in newborn rats and replaced the strip, reversing its orientation, in the experimental group. They found that the sutures behaved in a fashion similar to their new position in the skull and not to their origin. This indicates the dura as the controlling factor, not the suture. Although investigations into sutural biology continue to provide new insights, the relative importance of the lack of stretch across the suture and the relationship of the dura to the suture remains unclear.
Biology and Genetics
Overall incidence of craniosynostoses is estimated at 1 in 2000-3000 births in the United States.
Biology
An important distinction must be made between sporadic (nonsyndromic) craniosynostoses and syndromic synostoses. Sporadic synostoses generally are isolated, whereas syndromic synostoses include a variety of abnormalities often related to limb formation and facial development. The physiologic processes responsible for these aberrations of normal development are quite different.
Bresnick and Schendel compared fibroblast growth factor receptor (FGFR)-2 activity in stenosed and nonstenosed sutures in patients with Crouzon syndrome, Apert syndrome, and nonsyndromic coronal synostosis. Their findings demonstrate a lower level of activity in the stenosed sutures of patients with Crouzon and Apert syndrome compared to patients with nonstenosed sutures. However, no difference was observed in sutures of patients with nonsyndromic coronal synostosis. This data argues for different mechanisms in sporadic versus syndromic synostoses. A difference in mechanisms may indicate a difference in the genetic locus responsible for the abnormality. In syndromic synostoses, the exact gene containing the mutation responsible for the disease has, in most cases, been identified. The genes responsible for nonsyndromic cases have not yet been elucidated and may be found in different loci from those for syndromic synostoses.
Regardless of sporadic or syndromic type, current belief holds that the physiologic cause for premature closure of sutures lies in the biology of the dura, not solely in the osteogenic front of the sutures.
Opperman showed that removing the dura underlying the coronal sutures of a fetal rat reliably results in fusion of that suture. This dura-dependent patency was attributed to the presence of heparin-binding factors. In a later experiment, the same team found that manipulation of the overlying periosteum does not affect suture patency.
Further work suggests that the heparin-binding substances may be transforming growth factor (TGF)-beta 1, TGF-beta 2, and TGF-beta 3. Specifically, an experimental model of coronal synostosis (involving removal of dura) was studied in comparison to normal coronal sutures (with dura left undisturbed). Increased expression of TGF-beta 1 and TGF-beta 2 was observed in the fusing coronal sutures, whereas increased levels of TGF-beta 3 were associated with the patent coronal sutures.
Similarly, Lin demonstrated increased TGF-beta 2 and TGF-beta 3 in stenosed sutures of patients with persistent lambdoid synostosis. Roth studied sporadic cranial synostosis and found increases in TGF-beta 1 and TGF-beta 2 in stenosed sutures; however, consistent with Opperman's findings, TGF-beta 3 was associated with patent sutures. Longaker found a level of TGF-beta 1 to be 26 times greater in a fusing suture than in the control. TGF-beta 1 induces the production of basic fibroblast growth factor (bFGF), which causes an increase in collagen production and mineralized bonelike nodules. This may be an important part of the process of premature suture fusion.
In a series of experiments, Longaker examined the role of the dura in sutural patency and the specific molecular signals involved in this process. [9] In the adult rat the posterior frontal suture normally fuses at 12-22 days. All other sutures remain patent throughout the animal's life. Sutural fusion is greatly delayed when the dura is separated from the posterior frontal suture with an impermeable membrane.
The importance of the regional function of the dura was demonstrated through an experiment in which the posterior frontal and sagittal sutures were removed as an intact strip, reversed in position, and placed back on the cranium. The sutural fate reversed, and the previously posterior frontal suture (now located over the sagittal dura) remained patent while the sagittal suture (now overlying the posterior frontal dura) fused. These findings were confirmed in vitro, thus removing the influence of the forces exerted by variable growth of the skull and cranial base. Furthermore, when the suture is isolated from the underlying dura mater, in vitro suture fusion does not occur, demonstrating the importance of the dura for normal cranial development.
Hobar found that dura mater from neonatal guinea pigs, when transplanted into an area of cranial defect, closes that defect. Dura from an adult guinea pig is not capable of this healing. Longaker demonstrated the ability of immature dura (compared to adult dura) to induce elevation in the production of TGF-beta 1, TGF-beta 3, osteocalcin, and collagen types 1 and 3. These data indicate the healing potential of immature dura and may explain how large calvarial deficits are closed in children. Current investigations are exploring the potential use of this phenomenon through genetic manipulation of mature dura to induce a more immature function.
Searching for molecular factors responsible for suture control, Longaker studied insulinlike growth factor (IGF) in the fusing sutures of rats. When compared to patent sutures, IGF-I and IGF-II expression significantly increased in the dura underlying the posterior frontal suture approximately 1 week prior to sutural fusion. Interestingly, a similar elevation was observed in connective tissue from the osteogenic front of the posterior frontal suture during fusion. IGF increases the production of osteocalcin, which is found in high levels of closing sutures.
In studies comparing the fusing sutures of patients with unilateral coronal synostosis (UCS) with normal, nonfusing sutures, a cDNA fragment was identified as increased in the fusing sutures. The gene for this fragment is NELL-1. It is expressed preferentially in intramembranous bone and neural tissue. The exact role of this gene is still unknown. As FGF is involved in the process of suture fusion, Delezoide studied the expression of FGF receptors in human bony development. Results demonstrated an increase in the expression of FGFR-1 and FGFR-2 in limb development. Cranial development was associated with an increase in the expression of FRFR-1, FGFR-2, and FGFR-3. These findings make sense when one considers the genetics of craniosynostoses. [10]
Genetics
The genes responsible for synostotic syndromes are related to the molecular factors involved in cranial suture biology. TGF-beta and IGF molecules are the most abundant growth factors found in bone. FGFR molecules also play an important role in cranial development. Logically, the genes encoding these substances are the most likely loci associated with craniosynostoses.
Beginning in 1987, researchers elucidated the chromosomes containing specific mutations responsible for various syndromes. Stickler syndrome, located on chromosome 12, was the first syndrome related to a specific chromosome. This condition is associated with myopia, vitreoretinal degeneration, cleft palate, and arthropathy. Next, Van Der Woude syndrome was discovered to be located on chromosome 1. Further work revealed the locations of Treacher Collins syndrome on chromosome 5 and Saethre Chotzen syndrome on chromosome 7.
In 1991 the first gene associated with craniosynostosis, GLI-3, was identified. It is associated with Greig cephalopolysyndactyly, which is occasionally associated with craniosynostosis. In 1993, Jabs identified the first gene responsible for a specific craniosynostosis syndrome, Boston-type craniosynostosis, which involves a variety of abnormal head shapes. Warman reported a series of 19 patients with this condition, leading to the identification of the gene's location on chromosome 5 and shortly thereafter to the identification of the specific gene, MSX2.
FGF receptors may be involved in the pathogenesis of many syndromic craniosynostoses. Research into the genetic origin of these molecules has led to the discovery of many loci related to syndromic craniosynostoses (eg, Crouzon syndrome, Apert syndrome). These syndromes are related to mutations in specific genes encoding various FGF receptors, specifically, FGFR-1 on chromosome 8, FGFR-2 on chromosome 10, and FGFR-3 on chromosome 4. The greatest number of mutations identified as causes of craniosynostoses are found on chromosome 10 and are related to FGFR-2. The large variety of known mutations is listed below.
Some observations regarding mutations causing craniosynostoses are important. Historically, researchers believed that a single mutation causes a single disease. Current knowledge disproves this notion. The same mutation in chromosome 10 (Cys342Arg) in exon 9 has been observed in patients with Crouzon syndrome as well as Apert syndrome. That is, the same mutation has caused a different syndrome in different families. However, within the same family, the phenotype breeds true.
One explanation offered for this phenomenon is that sequence polymorphisms within the mutated gene (which can vary among people) may affect the expression of that gene, resulting in different phenotypes despite identical mutations. There are also cases of the same mutation within a given family resulting in different syndromes. The sequence polymorphism theory also may explain this situation.
Interestingly, Tessier alluded to the close relationship between patients with Crouzon syndrome and those with Apert syndrome as early as 1986 when he defined a transitional phenotype between the typical Crouzon and Apert manifestations, dubbed "Croupert." [11]
While there may be a variety of phenotypes resulting from the same mutation, generally the relationship of mutations to syndromes is well conserved. Apert syndrome, for example, has been attributed to one of two specific mutations in most patients (chromosome 10, Ser252Trp or Pro253Arg). In some cases a host of different mutations in different patients cause the same syndromes. This is true of Crouzon syndrome and particularly of Pfeiffer syndrome, which can result from mutations in FGFR-1 on chromosome 10 or FGFR-2 on chromosome 4. Research continues to identify new mutations related to syndromic craniosynostoses. For example, the gene for Jacobsen syndrome, manifested by trigonocephaly, facial dysmorphism, cardiac and hematologic defects, and intellectual disability has been located on chromosome 11q25. The exact locus for mutations causing Saethre-Chotzen syndrome has been found on chromosome 7 and termed the TWIST gene.
As genetic links to disease are elucidated, the potential for exact diagnosis increases. Currently, detection is possible for many mutations causing Apert, Crouzon, Jackson-Weiss, and Saethre-Chotzen syndromes with varying accuracy. With improving techniques in molecular biology, it is not unreasonable to anticipate the discovery of genetic therapies to aid or cure syndromic dysostoses.
Diagnosis and Treatment
The following terms describe premature fusion of the following sutures:
-
One coronal suture - Plagiocephaly (see images below; often used as generic term; literally, "crooked head")
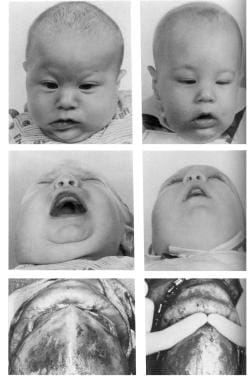
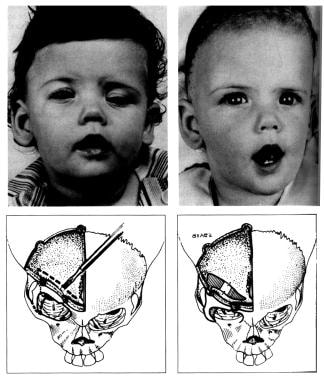 Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. This child was not treated until 10 months of age, which is older than one would prefer. The right coronal suture was released, and the right supraorbital rim and orbital roof along with the lateral orbital rim down to the takeoff of the zygomatic arch were advanced about 2 cm, with a greenstick fracture in the nasofrontal region. A small strut of bone was placed in the gap in the orbital roof to maintain the advancement, and the frontal bone was advanced to the supraorbital rim without being turned. Note on the postoperative photographs that the left brow is slightly lower than the right. Remember to remove a bit of scalp on the unaffected side to give symmetry of the brows.
Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. This child was not treated until 10 months of age, which is older than one would prefer. The right coronal suture was released, and the right supraorbital rim and orbital roof along with the lateral orbital rim down to the takeoff of the zygomatic arch were advanced about 2 cm, with a greenstick fracture in the nasofrontal region. A small strut of bone was placed in the gap in the orbital roof to maintain the advancement, and the frontal bone was advanced to the supraorbital rim without being turned. Note on the postoperative photographs that the left brow is slightly lower than the right. Remember to remove a bit of scalp on the unaffected side to give symmetry of the brows.
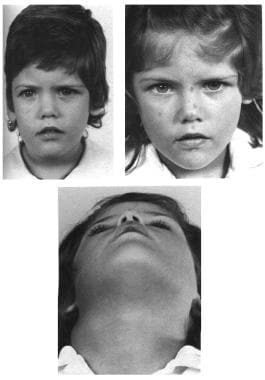 Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. With growth the correction maintains itself, as one can see in the photographs at age 3 and 6 years.
Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. With growth the correction maintains itself, as one can see in the photographs at age 3 and 6 years.
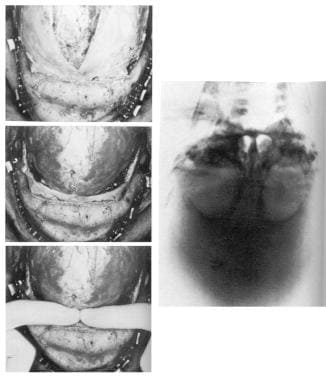
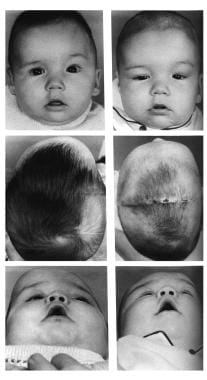 Congenital, synostoses. Plagiocephaly due to left hemicoronal synostosis. Release of the left coronal suture had been performed at 2 weeks of age because of ventricular displacement and signs of increase intracranial pressure. At age 5 months, retrusion of the left supraorbital region and frontal bone were still evident. The preoperative radiograph showed a pronounced "harlequin" orbit due to displacement of the sphenoid ridge. Here the supraorbital bandeau orbital roof complex was taken out in one piece, advanced on the left, and recessed slightly on the right; the frontal bone segments were exchanged to provide a symmetrical forehead. Some scalp had to be excised on the unaffected side to give brow symmetry.
Congenital, synostoses. Plagiocephaly due to left hemicoronal synostosis. Release of the left coronal suture had been performed at 2 weeks of age because of ventricular displacement and signs of increase intracranial pressure. At age 5 months, retrusion of the left supraorbital region and frontal bone were still evident. The preoperative radiograph showed a pronounced "harlequin" orbit due to displacement of the sphenoid ridge. Here the supraorbital bandeau orbital roof complex was taken out in one piece, advanced on the left, and recessed slightly on the right; the frontal bone segments were exchanged to provide a symmetrical forehead. Some scalp had to be excised on the unaffected side to give brow symmetry.
-
Both coronal sutures - Brachycephaly (see image below)
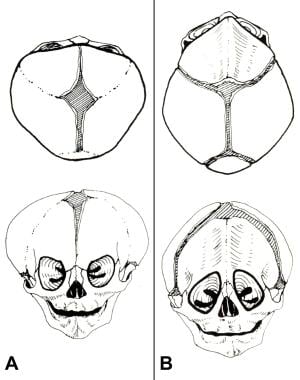 Congenital, synostoses. (A) In brachycephaly, both coronal sutures fuse prematurely, producing a broad short head and often some degree of orbital hypertelorism. (B) In trigonocephaly, the metopic suture fuses prematurely, producing a keel-shaped frontal ridge and often a mild hypotelorism.
Congenital, synostoses. (A) In brachycephaly, both coronal sutures fuse prematurely, producing a broad short head and often some degree of orbital hypertelorism. (B) In trigonocephaly, the metopic suture fuses prematurely, producing a keel-shaped frontal ridge and often a mild hypotelorism.
-
Metopic suture - Trigonocephaly (see images above and below)
 Congenital, synostoses. Trigonocephaly. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline. The deformity is rarely seen in adults, suggesting that it tends to be self-correcting without surgical intervention.
Congenital, synostoses. Trigonocephaly. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline. The deformity is rarely seen in adults, suggesting that it tends to be self-correcting without surgical intervention.
 Congenital, synostoses. Trigonocephaly. The preoperative radiograph has a hypoteloric appearance. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline.
Congenital, synostoses. Trigonocephaly. The preoperative radiograph has a hypoteloric appearance. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline.
-
Sagittal suture - Scaphocephaly (see image below)
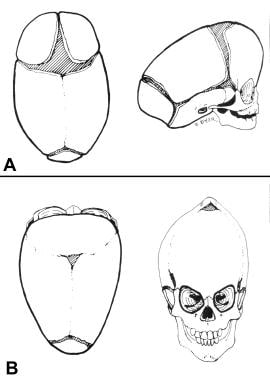 Congential, synostoses. (A) Premature fusion of the sagittal suture produces scaphocephaly. The head is narrow and long. The forehead may be somewhat protrusive, but few other facial manifestations occur. (B) Premature fusion of all the cranial sutures causes turricephaly. The skull has nowhere to grow but up.
Congential, synostoses. (A) Premature fusion of the sagittal suture produces scaphocephaly. The head is narrow and long. The forehead may be somewhat protrusive, but few other facial manifestations occur. (B) Premature fusion of all the cranial sutures causes turricephaly. The skull has nowhere to grow but up.
-
Lambdoid suture - Occipital plagiocephaly
-
Multiple cranial sutures - Acrocephaly, turricephaly (see image above), oxycephaly
-
Kleeblattschädel anomaly (all with different phenotypic appearances)







Syndromic dysostoses are associated with a variety of abnormal skull shapes. Although some syndromes typically occur with certain synostotic sutures, a wide range of combinations exists. Syndromic dysostoses are confounded by forces of growth in an abnormal facial skeleton. Varying relationships of syndrome and synostosis result.
Radiology
Plain skull radiographs were the first method used to radiologically evaluate patients with suspected craniosynostosis. At least 4 views are necessary to examine all sutures. If one suture in particular raises concern, a tangential view of this area is useful, particularly if a bony ridge is palpable. Even a small segment of fused suture may profoundly delay or arrest growth. Because of this, multiple views are required to completely evaluate the skull for suture synostosis.
Historically, the next modality used to diagnose craniosynostosis was bone scintigraphy. Early diagnosis (prior to closure of the suture) relies upon heightened activity along the suture line, while later diagnosis (after closure) is supported by a decrease in normal activity. In cases that seem equivocal by plain film, scintigraphy can add useful information and aid in diagnosis. Overall, scintigraphy is not as reliable as plain film for the evaluation of craniosynostosis.
The advent of CT revolutionized the diagnosis of fused cranial sutures. Closed sutures appear sclerosed or obliterated on CT bone windows; defects in parenchyma and ventricle spaces can be evaluated on soft tissue windows. Three-dimensional rendering of CT data provides a model of the cranial vault including a depiction of the patient's sutures and fontanelles, and accuracy is improving with newer generations of CT software. At most, obtain 1.5 -mm cuts to allow 3-D reconstructions of acceptable quality. CT technology has obviated the need for plain films, though radiographs remain an option in facilities lacking CT capability.
Sonograms have been used more recently to evaluate cranial sutures. Patency of sutures can be evaluated transcutaneously without the use of ionizing radiation. In patients in whom the diagnosis of craniosynostosis is in question, sonography may prove ideal. Prenatal diagnosis may become common and is already being tested in animal models. The advantage of such early diagnosis is questionable as prenatal intervention for a nonfatal condition is not advisable. Until techniques are developed to allow safe prenatal treatment of craniosynostosis, CT remains the diagnostic criterion standard.
Historic Treatment
In 1890, Lannelongue reported on craniectomy performed on patients with microcephaly in an attempt to prevent imbecility. [12] The report prompted Cushing's acerbic commentary from "Insanity and Imbecility."
It is unaccountable that an idea should have arisen attributing microcephalus and other conditions of congenital imperfection, as well as those of development arrested by disease, to a premature closure of the cranial bones. All of our knowledge goes to show that an early closure of fontanel and suture is due to a primary failure of growth of the encephalon; not to the reverse, a failure of growth due to a primary closure.
Experiments by d'Abundo and others show that animals whose skulls have been firmly enclosed in early life so that no possibility of cerebral expansion exists cannot survive. Even firmly closed sutures may give way before the pressure of growth. This is observed even in young adult life as a separation of sutures in acquired hydrocephalus and in cases of tumor growth. The introduction in 1891 of linear craniectomy, which has led to innumerable operations allegedly followed by an improvement in mentality, is a lamentable instance of the furor operandi running away with surgical judgment. Addressing this, Jacobi's address, "Non Nocere," was delivered in 1894 before the International Medical Congress in Rome.
Educational rather than surgical or medicinal measures are indicated in these unfortunate cases. There has been a high mortality rate in these operations, and though death cannot be lamented, the surgeon is not a barbarian to execute the helpless.
Cushing was correct to criticize surgery for microcephaly associated with a primary lack of cranial growth potential but he was in error concerning the effects of premature sutural closure in infants with a healthy potential for cranial growth. Later, Faber and Towne, Dandy, and Ingraham et al showed that craniosynostectomy indeed was indicated to release prematurely fused sutures. [13, 14, 15]
Initially, the purpose of these procedures was to prevent development of increased intracranial pressure and subsequent neurologic damage. It was a controversial procedure at the time as it was believed that increased intracranial pressure would develop only if all of the sutures were fused, without possibility of compensatory growth in another direction. A study by Bantu on patients who did not undergo operations did not detect measurable neurologic damage. The debate raged in the neurosurgical literature as to whether any neurosurgical procedure could be recommended for "purely cosmetic purposes."
After Ingraham and Matson and Shillito and Matson reported a low mortality rate, isolated suture release became an accepted procedure to prevent subsequent deformity, even when increased intracranial pressure was not anticipated. [16, 17] Studies by Renier et al and Carmel et al show that even with premature fusion of one hemicoronal suture, increased intracranial pressure and ventricular deformity can occur. [18, 19, 20]
However, in some patients, craniostenosis was actually craniofaciostenosis. Despite adequate release of involved cranial sutures, an untreated stenosis of the "facial" (cranial base) sutures remained. In these situations, the patients developed significant facial deformities.
Hoffman, Mohr, and Bludell, influenced by Tessier's earlier work on adults, developed a "lateral canthal advancement" to be performed in conjunction with an extended release of the involved hemicoronal suture, carried into the inferior orbital fissure. [21, 22] This procedure consisted of advancement of the supraorbital rim and frontal bone and constituted the first attempt to place involved cranial and orbital segments into proper position at the time of operation, rather than relying on the growth of underlying brain and eye to correct the deformity after suture release.
For conditions involving accessible cranial and facial sutures (ie, plagiocephaly, pure brachycephaly, scaphocephaly, trigonocephaly), suture release and repositioning of deformed parts seems to correct the condition permanently. The correction is maintained even with further facial and cranial growth. For other conditions, notably the craniofacial dysostoses of Crouzon and Apert, opening the accessible sutures and repositioning the upper orbital and frontal bones are not corrective. Whether other cranial base sutures remain unreleased after surgery or the potential for facial growth simply is not present is uncertain. Those performing this type of craniectomy and/or repositioning for craniofaciostenosis (eg, Crouzon, Apert, Carpenter, Pfieffer) believe that although the subsequent orbital deformity is lessened, the lower maxillary deformity (as evidenced by anterior crossbite and class 3 malocclusion) develops unabated.
Surgical Treatment
Interceptive surgery
Some neurosurgeons prefer to operate on craniostenosis as soon as the diagnosis is established, which may be within the first month of life. At this age the frontal and orbital bones are little more substantial than cardboard and can be cut with a scissor rather than a power saw. However, they are so pliable that rigid fixation in an advanced (ie, proper) position is difficult.
Marchac and Renier coined the term "floating forehead," and it seems logical at this age simply to replace the released segments without fixation and to rely on subsequent brain growth. [23] Use fixation when bones are solid enough to be maintained in an advanced position (4-6 mo or older). In patients older than 12 months, neurosurgeons are less optimistic about the results of craniosynostectomy. The individual neurosurgeon decides whether to put a polyethylene or Silastic sheet over the edges of the released suture in the hope of preventing reossification. The authors' impression is that reossification will occur despite the foreign body, both over and under it, because the major osteoprogenitor layers are the dura and the periosteum, not the cut edges of bone.
This early interceptive surgery does not preclude the need for a subsequent corrective operation. However, as it does seem to lessen the severity of the orbital deformity in cases of coronal/metopic synostosis, it may mean that a lesser operation – a subcranial LeFort III rather than a monobloc, or a LeFort II rather than a LeFort III – is required. The neurosurgeon performs the suture release anyway, and performing an advancement of the orbital roof, supraorbital bandeau, and frontal bone does not lengthen the procedure excessively, nor does it interfere with a later, more definitive operation if the bony orbital framework is properly reconstituted.
Current surgical treatment
One should differentiate the pure craniosynostoses, which are largely nonsyndromic, from the craniofacial dysostoses, such as Crouzon syndrome, Apert syndrome, and other named conditions (eg, Pfeiffer, Carpenter, Saethre-Chotzen). These later conditions generally are Mendelian dominant, and for most, the genetic DNA abnormality has been delineated precisely. Since the craniosynostoses involve only cranial sutures, if the prematurely closed suture(s) is released and abnormally positioned cranial and orbital bone segments properly positioned, the correction should be maintained with growth of the brain, and further surgery is rarely required. In these conditions the facial sutures are normal and facial growth should be normal.
In contrast, in the craniofacial dysostoses facial sutures are not normal, and release of a prematurely fused cranial suture will not bring about normal facial growth. Patients with the syndrome, although they may be treated at their first operation similarly to those with craniosynostoses, usually require further operations during the first 2 decades of life to correct functional and morphologic abnormalities resulting from lack of growth of facial sutures. For example, a patient with brachycephaly may be indistinguishable from a patient with Crouzon syndrome at birth, and both may be treated by fronto-orbital advancement at 6 months. The child with brachycephaly likely needs no further surgery; the child with Crouzon syndrome will most likely develop exorbitism and retromaxillism because of impaired midfacial growth, which requires further surgical treatment.
Timing of surgery can vary depending on the type of craniosynostosis and its severity. If pansynostosis is present, a surgical emergency exists since brain growth is not possible, and the operation may need to be repeated at intervals if reossification occurs, limiting cranial growth.
A child with significant scaphocephaly may be operated on at 3-4 months, but for most other conditions a better morphological result can be obtained by waiting until 6-8 months. The calvarial and orbital bone is better formed at this age and can be shaped and fixed with more precision and stability. The goals of surgery are to release the prematurely closed suture(s) and to reshape abnormally positioned bones so that they are in a proper position and symmetric with the contralateral side.
New approaches have been described in the literature, particularly in cases recognized and diagnosed early. The use of springs, described by Lauritzen et al, has compared favorably with the standard craniofacial procedures for the correction of cranial synostosis. [24] Barone and Jimenez have reported successful results with early treatment of infants with cranial synostosis using endoscopic-assisted minimally invasive suturectomies followed by the use of custom-made cranial helmets. [25] The determining factors in having adequate outcomes are early diagnosis and early surgical treatment. The benefits of this approach are many, including less invasive surgery with minimal scars, less operating and hospitalization time, less need for blood transfusion, and overall earlier recovery.
The series of images below show a child whose severe scaphocephaly was successfully treated with a minimally invasive approach.
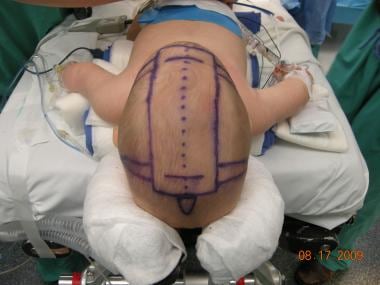 Preoperative picture of a 3-month-old child with a severe scaphocephaly. In this case, the authors used the minimally invasive approach with the aid of the laparoscope. The markings show the extent of the bone removed along the area of the sagittal suture (dotted line), the lateral barrel cuts, and the 2 incisions.
Preoperative picture of a 3-month-old child with a severe scaphocephaly. In this case, the authors used the minimally invasive approach with the aid of the laparoscope. The markings show the extent of the bone removed along the area of the sagittal suture (dotted line), the lateral barrel cuts, and the 2 incisions.
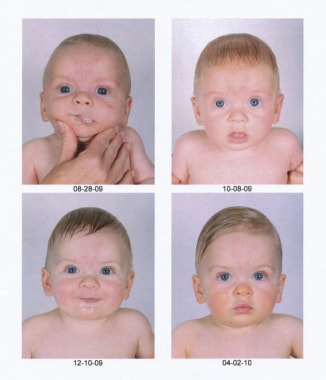 Postoperative pictures of the same child showing the impressive progressive improvement.
Postoperative pictures of the same child showing the impressive progressive improvement.
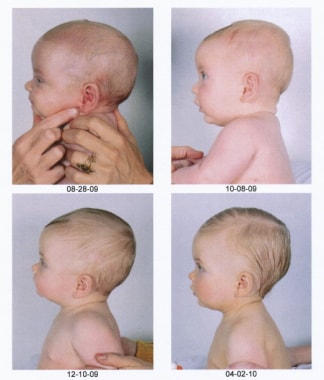 Postoperative pictures of the same child showing the impressive progressive improvement.
Postoperative pictures of the same child showing the impressive progressive improvement.
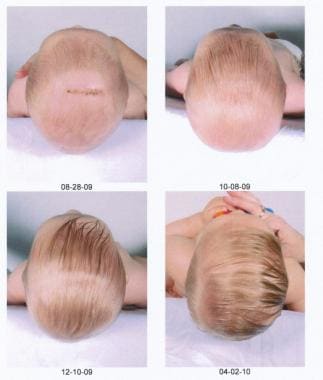 Postoperative pictures of the same child showing the impressive progressive improvement.
Postoperative pictures of the same child showing the impressive progressive improvement.
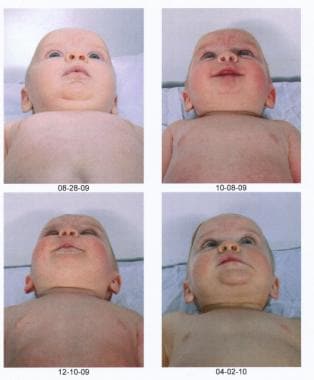 Postoperative pictures of the same child showing the impressive progressive improvement.
Postoperative pictures of the same child showing the impressive progressive improvement.
Incisions and dissection
A coronal incision too close to the anterior hairline yields a visible scar. Avoid this at all costs, since an unsightly scar in this area is impossible to improve and may be the only reminder of surgery if all else is corrected properly.
Begin the coronal incision in the sideburn above the ear and angle backwards at 45° for 3-5 cm, then cross the parietal bone to join the incision from the other side at the vertex. It is preferable to carry out the dissection in the supraperiosteal plane to just above the supraorbital ridges, where the dissection continues in a subperiosteal plane. The temporalis muscle may not be well developed but deserves careful respect. It can be reflected from the lateral orbital rim and temporal fossa and carefully repositioned at the end of the procedure.
In a reoperation where initial surgery was performed through an anteriorly placed incision (as described above), enter through the previous incision, since performing a more posterior incision will challenge the blood supply of the intervening scalp, occasionally resulting in a full-thickness loss of this tissue.
Technique
Perform the orbital and supraorbital osteotomies with reciprocating and oscillating saws with fine blades under constant irrigation. Use a high-speed craniotome with a fine cutting burr for the craniotomies.
For years, the authors' approach has been to provide acceptable stability with as little hardware as possible. Vicryl sutures are quite adequate for fixing cranial bone segments; use biodegradable plates and screws (as little as possible) when further stability is required. On rare occasions, one or two stainless steel wires can be used but try not to use any metallic material. Titanium microplates and microscrews were used briefly about 1 decade ago, but have been abandoned because they can translocate to an intracranial position with growth of the skull and were associated with minor localized growth disturbances that in a number of cases required further surgery.
Most children undergoing craniosynostosis surgery require blood, but there are a number of things that can be done to minimize blood loss. To diminish the need for intraoperative blood administration, perform infiltration along the coronal incision line with a 1:150,000 adrenaline/saline solution with hyaluronidase 10 minutes before making the incision; make an incision in the scalp very superficially with a scalpel and perform the rest of the incision and dissection with the Colorado tip on electrocautery; apply scalp clips, give intraoperative aprotinin, use hypotensive anesthesia during the coronal incision and its closure, and give preoperative erythropoietin. Craniostenosis procedures have been performed by the authors with total blood loss as low as 45 cc in a Jehovah's Witness to avoid transfusion, but always have blood available and parental permission for use should the situation become life threatening.
Aprotinin, an antifibrinolytic agent used to reduce operative blood loss in patients undergoing open heart surgery, is now only available via a limited-access protocol. Fergusson et al reported an increased risk for death compared with tranexamic acid or aminocaproic acid in high-risk cardiac surgery. [26]
Do not shave the head; at most, cut a strip about a centimeter in width and cover it with Tegaderm at the end of the procedure. Occasionally a small closed-system vacuum drain may be used. Light dressings are discretionary.
Unilateral coronal synostosis (UCS; plagiocephaly)
There is debate as to whether the fronto-orbital segment should be removed and repositioned on just the affected side or on both sides. Individualize treatment after a careful determination of the morphologic alterations in each particular patient. If there is severe deformity, it may be easier to obtain symmetry by reshaping the entire fronto-orbital segment. In older patients, it may be best to make an entirely new forehead from other areas of the skull; Marchac wire templates are quite useful when performing this procedure. If there is significant retrusion or protrusion of the temporal fossa on the affected side, correct this by advancing and bending the flat temporal bone or reversing a ballooning temporal bone. Usually a long tenon segment is extended from the lateral orbital rim, which is then fixed (after appropriate bending) to the intact temporal bone. A single biodegradable plate with 4 or 5 screws and an interpositional bone graft provide adequate stability.
Bone craftsmanship should be good enough that this segment is precisely symmetric with the contralateral normal side. Do not expect further growth of the brain to self-correct asymmetry. The contralateral normal eyebrow is often lower in UCS; remember to resect a bit of scalp on this side to level the eyebrows at the end of the procedure.
The nasal root is deviated to the side of the deformity, with the nasal tip pointing toward the normal side in UCS. A primary procedure to straighten the nose is not performed in infants. If the procedure is performed on a child aged 2-3 years and the nose is substantially deviated, perform an aligning osteotomy. Generally, it is better to wait and see if the nasal deviation corrects itself, which in the authors' experience usually occurs. In several patients in whom the nasal deviation persisted, monobloc nasal osteotomies were performed at 8 years when there was less concern for permanent tooth roots.
Bilateral coronal synostosis
The procedure is the same as that for UCS but is performed on both sides. Carry out an advancement of at least 15 mm at the temporal level, and advance the nasion straightforward, not cephalad.
Posterior positional plagiocephaly
Most cases with flattening of the occipital area, where the ipsilateral ear is positioned anteriorly (on the flat side), are positional deformational plagiocephaly, not true lambdoidal synostosis. [27] Treat most positional deformities by physical therapy for associated torticollis and possibly molding helmets, not surgery. If surgery is to be performed for a true lambdoid synostosis or a severe positional deformity that did not respond to helmet therapy, the procedure is similar to the frontal area procedure in coronal synostosis. Develop and advance an occipital bar, and attach the remaining occipital bone to the bar in a position of symmetry. Large venous structures – the torcula and lateral sinuses – are present beneath the occipital bone and should be carefully respected. Major bleeding from them can be fatal.
Trigonocephaly
Treatment of trigonocephaly is similar to bilateral coronal synostosis, but perform opening osteotomies along the fronto-orbital bar. The tenon coming off the lateral orbit should be wider and should extend far into the temporal bone to correct the temporal constriction that is usually associated with metopic synostosis. In mild cases, where there is only a ridge along the metopic suture but no keel-shaped deformity of the forehead, simply burr down the bony ridge or merely observe the patient and see if the condition improves with time.
Scaphocephaly
Milder cases have been treated with simple strip craniectomies with adequate results. Treat more severe cases with total cranial vault remodeling, removing and shortening a sagittal strip, rotating the occipital bone forward to shorten the anteroposterior dimension of the head, and repositioning more laterally the temporoparietal segments that are sectioned with barrel stave osteotomies, expanding the width of the head.
Recently, distraction osteogenesis has been employed to correct the cranial abnormalities associated with scaphocephaly. Distraction devices both increase the width of the cranial vault (intertemporal distance) and decrease anterior-posterior length. Distraction osteogenesis may prove useful in the treatment of other cranial dysostoses.
Turricephaly
Develop and shorten a biparietal bar and carry out a posterior expansion using the barrel stave procedure, along with a downward movement of the vertex. In some patients, cranial molding helmets used postoperatively help the skull develop in the desired directions and prevent undesirable head shapes.
Secondary surgery
This has become much less common with the avoidance of all metallic fixation devices. Some minor irregularities may be treated by limited burring or by addition of small bone grafts and pericranial flaps. In some cases of UCS the nose may require straightening. Widening of a coronal scar may require revision, which is not always successful. Artificial materials such as hydroxyapatite are not recommended for use in growing children but they can be used in adolescents or adults in limited amounts. It is best to use autogenous materials whenever possible.
Contraindications
The only absolute contraindication for this surgery is a medically unstable patient for whom other medical issues preclude this type of procedure.
-
Congenital, synostoses. Normal cranial suture anatomy at birth.
-
Congenital, synostoses. (A) In brachycephaly, both coronal sutures fuse prematurely, producing a broad short head and often some degree of orbital hypertelorism. (B) In trigonocephaly, the metopic suture fuses prematurely, producing a keel-shaped frontal ridge and often a mild hypotelorism.
-
Congential, synostoses. (A) Premature fusion of the sagittal suture produces scaphocephaly. The head is narrow and long. The forehead may be somewhat protrusive, but few other facial manifestations occur. (B) Premature fusion of all the cranial sutures causes turricephaly. The skull has nowhere to grow but up.
-
Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. This child was not treated until 10 months of age, which is older than one would prefer. The right coronal suture was released, and the right supraorbital rim and orbital roof along with the lateral orbital rim down to the takeoff of the zygomatic arch were advanced about 2 cm, with a greenstick fracture in the nasofrontal region. A small strut of bone was placed in the gap in the orbital roof to maintain the advancement, and the frontal bone was advanced to the supraorbital rim without being turned. Note on the postoperative photographs that the left brow is slightly lower than the right. Remember to remove a bit of scalp on the unaffected side to give symmetry of the brows.
-
Congenital, synostoses. Plagiocephaly due to right hemicoronal synostosis. With growth the correction maintains itself, as one can see in the photographs at age 3 and 6 years.
-
Congenital, synostoses. Trigonocephaly. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline. The deformity is rarely seen in adults, suggesting that it tends to be self-correcting without surgical intervention.
-
Congenital, synostoses. Trigonocephaly. The preoperative radiograph has a hypoteloric appearance. The supraorbital bandeau is removed, the lateral portions are advanced, and the frontal bone segments are replaced, avoiding a continuity of bone across the midline.
-
Congenital, synostoses. Plagiocephaly due to left hemicoronal synostosis. Release of the left coronal suture had been performed at 2 weeks of age because of ventricular displacement and signs of increase intracranial pressure. At age 5 months, retrusion of the left supraorbital region and frontal bone were still evident. The preoperative radiograph showed a pronounced "harlequin" orbit due to displacement of the sphenoid ridge. Here the supraorbital bandeau orbital roof complex was taken out in one piece, advanced on the left, and recessed slightly on the right; the frontal bone segments were exchanged to provide a symmetrical forehead. Some scalp had to be excised on the unaffected side to give brow symmetry.
-
Preoperative picture of a 3-month-old child with a severe scaphocephaly. In this case, the authors used the minimally invasive approach with the aid of the laparoscope. The markings show the extent of the bone removed along the area of the sagittal suture (dotted line), the lateral barrel cuts, and the 2 incisions.
-
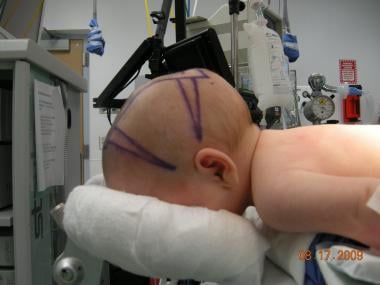
Preoperative lateral view of the child in the previous image.
-
Postoperative pictures of the same child showing the impressive progressive improvement.
-
Postoperative pictures of the same child showing the impressive progressive improvement.
-
Postoperative pictures of the same child showing the impressive progressive improvement.
-
Postoperative pictures of the same child showing the impressive progressive improvement.


