Background
Abnormalities of red cell membrane cation permeability are seen in several hereditary disorders. These dominantly inherited conditions are collectively called the hereditary stomatocytoses and allied disorders. This class of hemolytic anemias is clinically diverse. [1] See image below.
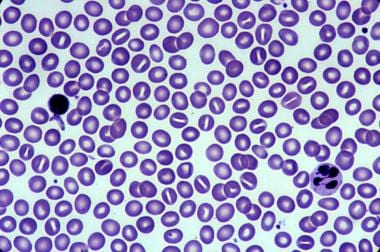 Hereditary stomatocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Hereditary stomatocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Overhydrated hereditary stomatocytosis (OHSt), also called hydrocytosis, was the first of these disorders to be described. [2] An abnormally increased cation influx results in swollen erythrocytes, hemolysis, and stomatocytes. At the other end of the spectrum, net loss of cations and water results in the more common dehydrated hereditary stomatocytosis (DHSt), also called xerocytosis.
Intermediate syndromes that are asymptomatic or have mixed features, such as cryohydrocytosis (CHC) and familial pseudohyperkalemia (FP), have also been described.
OHSt has been linked to mutations in the erythroid ammonia channel Rh-associated glycoprotein (RhAG), [3, 4] as well as, occasionally, to mutations in the band 3 chloride-bicarbonate exchanger AE1, regulated by SLC4A1. Decreases in stomatin are seen in OHSt. [5] The causative gene for DHSt was mapped to 16q23-24 and was identified with genome-wide sequencing as PIEZO1, with a few patients linked to KCCN4. [6] CHC has been linked to mutations in SLC4A1. [7] The causative gene for FP, mapped to 2q35-36, was identified as ABCB6.
Syndromic forms of hereditary stomatocytosis are rare but increasingly recognized and have improved our understanding of genetic mechanisms. Described syndromes including the following:
-
CHC with mental retardation, seizures, and hepatosplenomegaly (stomatin deficiency linked to SLC4A1)
-
Nonleaky stomatocytosis with hypercholesterolemia, xanthomas, splenomegaly, and macrothrombocytopenia (severe phytosterolemia linked to ABCG5 or ABCG8)
-
DHSt with transient perinatal edema, including neonatal ascites (linked to PIEZO1)
Pathophysiology
Erythrocytes have intracellular hemoglobin, 2-3,diphosphoglycerate (2,3-DPG), and ATP, which all exert osmotic pressure across the semipermeable cell membrane. By transporting Na+ and K+ ions across the cell membrane, red cells can adjust the intracellular concentration of these cations and regulate intracellular hydration. Any disturbances in membrane cation permeability alter cellular hydration and can cause numerous effects, including hemolysis.
In overhydrated hereditary stomatocytosis, the major defect is a marked asymmetrical increase in passive Na+ and K+ permeability. The influx of Na+ exceeds the loss of K+, causing a net influx of water, overhydration, and swelling. The resulting hydrocytosis leads to increased osmotic fragility and decreased deformability, with consequent hemolysis. This is primarily due to mutations of the Rh-associated glycoprotein gene (RHAG), [3] with decreases in stomatin, or protein 7.2b, due to a trafficking alteration. [5]
In contrast, the primary abnormality in DHSt is a change in the relative membrane permeability to K+. Efflux of K+ is increased 2- to 4-fold and results in cation depletion, with decreased intracellular osmolality and water loss. The xerocytes formed are shear-sensitive and prone to membrane fragmentation in response to metabolic stress, with subsequent hemolysis. DHSt has been mapped to gain-of-function mutations in PIEZO1 and KCCN4, with the latter affecting the Gardos channel. [6] FP has been linked to mutations in the ABCB6 gene and is usually asymptomatic or (rarely) shows mild macrocytosis. When erythrocytes are cooled to room temperature or lower (eg, after phlebotomy), the net K+ leak is greater than expected and results in factitious hyperkalemia. [8, 9]
CHC clinically manifests with mild hemolytic anemia and is remarkable for an abnormality of the band 3 chloride-bicarbonate exchanger AE1. [7] At low temperatures, the defective anion channel appears to allow a significant cation leak, and autohemolysis may be seen in vitro at 4ºC. The protein defect results from missense mutations in the SLC4A1 gene. [10]
Epidemiology
Frequency
International
These hereditary syndromes are extremely rare, and accurate data concerning their prevalence are lacking; however, overall they are thought to be 40-50 times less common than hereditary spherocytosis. [11] DHSt is reported in 1 per 50,000 live births. [12] and DHSt kindreds in Ireland [13] and France [14] have been reported.
Mortality/Morbidity
Morbidity in these disorders depends on the severity of the hemolytic anemia. The risks for neonatal hyperbilirubinemia with kernicterus are similar to those of other hemolytic anemias. Exchange transfusion is occasionally required. Aplastic crises associated with parvovirus infection occur, although they are infrequent. Both OHSt and DHSt are associated with a significant risk of serious thrombosis after splenectomy, although the reason for this is unknown.
Most patients with overhydrated hereditary stomatocytosis have chronic low-grade anemia punctuated by recurrent episodes of more severe anemia and jaundice. Other patients have a much milder disease. Iron overload, regardless of transfusion status, is now well recognized.
Most patients with dehydrated hereditary stomatocytosis are asymptomatic but experience mild-to-moderate hemolytic anemia, which is generally well compensated. Hydrops fetalis and neonatal ascites have been reported in a few kindreds. [15] Exchange transfusions are occasionally required. Even simple transfusions carry risks of infection, allergic reactions, and febrile or hemolytic transfusion reactions.
Race
Most patients with hereditary disorders of red cell permeability are of European descent.
Sex
These syndromes have no known sex predilection, and clinical manifestations are not affected by sex.
-
Hereditary stomatocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.